anemii2 dr. mahler
DESCRIPTION
curs 2 anemii mahlerTRANSCRIPT

Testele speciale în anemii
Asist.Univ.Dr.Mahler Beatrice
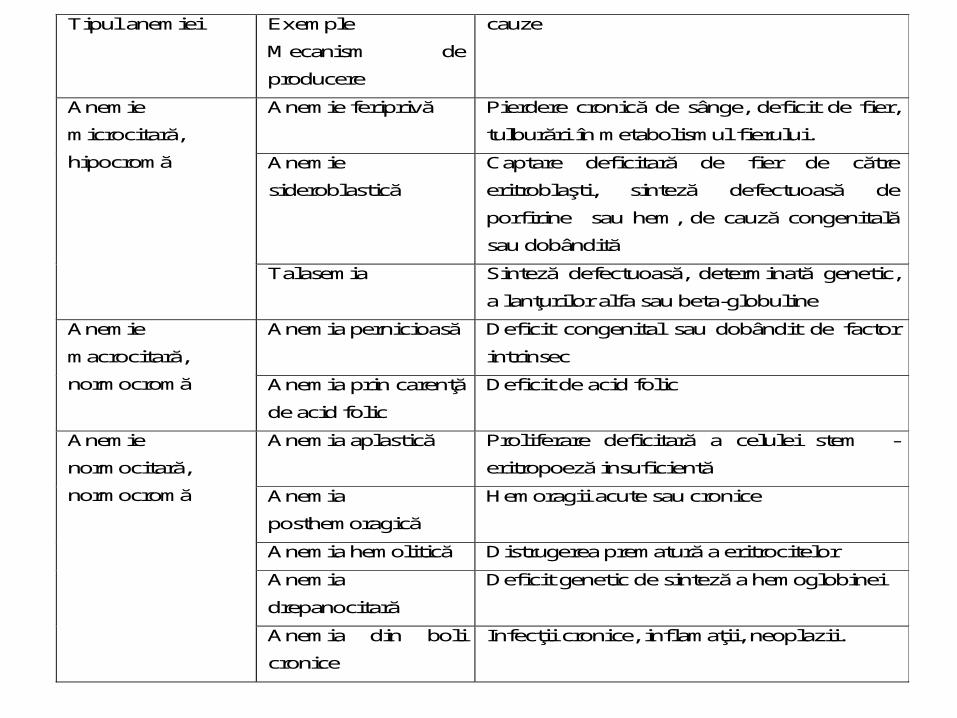
Tipul anemiei Exemple
Mecanism de
producere
cauze
Anemie
microcitară,
hipocromă
Anemie feriprivă
Pierdere cronică de sânge, deficit de fier,
tulburări în metabolismul fierului.
Anemie
sideroblastică
Captare deficitară de fier de către
eritroblaşti, sinteză defectuoasă de
porfirine sau hem, de cauză congenitală
sau dobândită
Talasemia
Sinteză defectuoasă, determinată genetic,
a lanţurilor alfa sau beta-globuline
Anemie
macrocitară,
normocromă
Anemia pernicioasă Deficit congenital sau dobândit de factor
intrinsec
Anemia prin carenţă
de acid folic
Deficit de acid folic
Anemie
normocitară,
normocromă
Anemia aplastică Proliferare deficitară a celulei stem -
eritropoeză insuficientă
Anemia
posthemoragică
Hemoragii acute sau cronice
Anemia hemolitică Distrugerea prematură a eritrocitelor
Anemia
drepanocitară
Deficit genetic de sinteză a hemoglobinei
Anemia din boli
cronice
Infecţii cronice, inflamaţii, neoplazii.

Anemii microcitare• 1. deficit de metabolizare a Fe
– An. Feriprivă –cauze- deficit de aport, malabsorbție, pierdere de fe, creșterea necesarului de Fe
– An. din boli cronice– Hipotransferinemie ereditară– An congenitală hipocromă-microcitară, cu încărcare de Fe –sdr.
Shahidi – Nathan- Diamond• 2. porfiria și tulburări în sinteza Hem
– An. Sideroblastică • dobândită, idiopatică, • ereditată, transmitere X-linkată
• 3. tulburări de sinteză a Hb– Talasemia– Alte hemoglobinopatii – Hb instabile

Anemii normocorme, normocitare
• 1. cu răspuns medular bun– An. Posthemoragică– An. Hemolitică – fals macrocitară,,când cresc
reticulocitele • 2. cu răspuns medular deficitar– Hipoplazie medulară – an. Aplastică– Infiltratie medulară –metastaze medulare, mielofibroze– Scade producția de eritropoetină – afecțiuni renale, boli
cronice, malnutriție, afecțiuni hepatice,

An macrocitară• 1. deficit de vitamina B12
– Scăderea aportului alimentar– Scăderea absorbției – scade Factorul intrinsec, gastrectomii, malabsorbție, af. Intestinale
inflamatorii,– Parazitoze cu rol ompetitiv de absorbție – diphylobotrium latum– Necesar crescut – afecțiuni pancreatice cronice, sarcină, boli neoplazice, hipertiroidism,– Scăderea utilizării – deficit enzimatic
• 2. deficit de acid folic– Scade aportul , absorbția– Crește necesarul-sarcină, hipertiroidism, boli neoplazice,– Utilizare cu agoniști – metotrexat, trimetoprim– Pierdere- hemodializă
• 3. inhibitori metabolici – lipsa de răspuns la inhibitorii metabolici– Sinteza de purine- azatioprină– Sinteza de pirimidină– Sinteză de thymidylate– Sinteză de hidroxiuree- deficit sever de Fe,– Af. Congenitală – aciduria orotică congenitală,

Teste specifice în anemia hipocromă, microcitară
• Teste pentru diagnosticul deficitului de fier

Fierul
• Organismul conține 4,5 g Fe, repartizat în 3 sectoare:– -Fe hemoglobinic –72% - din circulație - legat de transferină– -Fe din depozite – 25% - feritină și hemosiderină – primele afectate– -Fe din comp. Enzimelor –citocrom, mioglobină, etc.
Metabolismul Fierului• Presupune 4 etape:• a.Aport şi absorbţie – absorbția fe se realizează la nivelul duodenului proximal, în
stomac este legat de proteine ( Fe feric), sub influența vit.C și HCl se desface în Fe feros este ulterior absorbit la nivelul enterocitului legat de apoferitină( Fe feric).
• b.Transportul Fe este realizat de transferină ( o beta globulină plasmatică sintetizată în ficat). Doar 1/3 din transferină leagă Fe, molecula prezentând 2 locusuri
• c.Utilizarea Fe – în sinteza hemoglobinei, mioglobinei• d.Reutilizarea Fe – eritrocitul are o durată de viață de aproximativ 120 zile, după
care este catabolizat în splină eliberând Fe, care este reutilizat, iar hemul este transformat în bilirubină. Globina este folosită la sinteza proteinelor.

Tulburarea metabolismului Fe
• a.Tulburarea aportului şi absorbţiei – scăderea aportului sau un necesar crescut (femei, copii), det. apariția anemiei. Ex .alcoolici, rezecții gastrice/ intest, gastro/enteropatii,
Ex. Anemii Sideropenice- Fe scăzut
• b.Tulburarea transportului – – apare în hipotransferinemii prin - def. de sinteză congenital
sau dobândit– catabolism excesiv –în inflamații cr sau ac(sdr. nefrotic)
ex. Anemii sideropenice – Fe scăzut

Metabolismul Fec.Tulburarea utilizării • creşte utilizarea Fe extramedular – în inflamații. cr. Crește
sinteză enzimelor neheminice, nelizozomale.• scade utilizarea Fe în compartimentul medular –deficit de
sinteză a hemului –def. enzimatic (hemsintetază) sau tulburări în funcția mitocondrială. = An sideroblastice sideroacrestice
ex, Anemii sideroblastice – fe crescutd.Tulburarea reutilizării – pierderi cr. de sânge• Ex. Anemii sideropenice – Fe scăzut

sideremia• Sideremia sau Fierul = 50 – 170 µg/dl• Interpretare• Scăderea nivelului de Fe :
– anemie prin deficit de fier,– pierdere cronică de sânge,– boli cronice ( boli infectioase, boli autoimune),– trimetrul III de sarcină,– absorbtie deficitară,
• Creşterea nivelului de Fe : – sindrom de supraabsorbţie a Fe,– anemie hemolitică,– tansfuzii multiple,– tratament excesiv cu Fe– hepatita acută– leucemie acută– deficit de vitamina B6.
• Valorile între 280-2550 µg/dl determina simptome de intoxicare Fe – dureri abdominare, greturi, varsaturi, diaree hemoragică, cianoză şi convulsii. Decesul survine la valori peste 1800 µg/dl
• Factorii care interfera cu– scădere Fe - etanol, estrogen, contraceptive orale,– creşterea Fe – antibiotice, aspirina si testosteronului.
• Determinarea sideremiei, fără transferină sau capacitatea totală de legare a fierului ( CTLFe) – este o evaluare limitata a Fe, cu excepţia otrăvirii cu Fe.

Transferina
• este o proteina de transport, sintetizată în cantitate mare în ficat, reglată de nivelul de absorbţie al fierului.
• Transferina este saturată cu Fe în proporţie de 33%.• Nivelul crescut al transferinei în sânge ajută organismul în lupta sa
antiinfectioasă.
• Transferina = 250 – 425 mg/dl
Interpretare• Scăderea transferinei :
– anemia microcitară din boli cronice,– deficit de proteine ( arşi, malnutriţi )– boli infecţioase cronice,– deficit genetic – atransferinemie ereditară,– boli hepatice acute şi renale cronice,
• Creşterea transferinei :– anemie prin deficit de fier– sarcină– tratament cu estrogeni.

Saturaţia transferinei
• este un index care evalueaza saturatia cu Fe. • Se calculeaza cu formula :
Saturatia transferinei = Fe2+ ( ionii ) x 100 CTLFe• Saturatia transferinei = 10 – 50 %.
• Interpretare • Creşterea saturaţie transferinei
– hemocromatoză– talasemie– hemosideroza– boli hepatice acute
• Scăderea saturaţie transferinei– anemia feriprivă– malignităţi– anemia din boli cronice

Capacitatea totală de legare a fierului (TIBC)
• este corelată cu nivelul transferinei, dar relaţia nu este liniară. Reprezintă cantitatea de Fe pe care ar transporta-o transferina, dacă ar fi saturată 100% cu Fe.
CTLFe = 250 – 450 µg/dl
• Interpretare:• Scăderea CTLFe :
– hipoproteinemii,– hemocromatoza,– anemia din boli cronice,– ciroza hepatică,– talasemie,– hipertiroidism,– boli renale cronice,
• Creşterea CTLFe :– deficit de Fe– sarcina în trimestrul III,– pierdere de sânge acută sau cronică,– hepatita acută.

Determinarea depozitelor de FeFeritina
• complex format din ioni de Fe 2+ şi o proteină – apoferitina, originară la nivelul
sistemului reticuloendotelial.
• Feritina reflectă depozitul de Fe şi este un indicator fidel al statutul total al Fe.
• Feritina, un complex ce contine hidroxid feric ( Fe2+ ) şi o proteină, APOFERITINA,
care provine din sistemul reticuloendotelial.
• Feritina reflectă rezerva de fier din organism şi este cel mai de încredere indicator al
nivelului total de Fe din organism. Examinarea măduvei osoase este cu siguranţă cel
mai bun test. Aspirarea măduvei osoase poate fi necesară în anumite situaţii, atunci
când are valoare normală la limita inferioară asemănătoare cu fierul seric scăzut în
anemia din bolile cronice.
• Determinarea feritinei este mai sensibilă şi mai specifică decât determinarea
concentraţiei Fe sau CTLFe pentru diagnosticarea deficitului de fier. Feritina începe să
scadă înaintea apariţiei anemiei şi a altor modificări.

Determinarea depozitelor de FeFeritina
• Valori Normale:• Bărbaţi : 20 – 250 µg/L, anemia din bolile cronice < 100 µg/L, iar în absenţa inflamaţiei < 20 µg/L.• Femei : 10 – 120 µg/L, anemia din bolile cronice < 20 µg/L, iar în absenţa inflamaţiei < 10 µg/L.• Copii : 7 – 140 µg/L. Nou-născuţii : 25 – 200 µg/L. ↓
Interpretare : • Scăderea feritinei ( <10 µg/L ), indicator uzual in anemia prin deficit de fier.
– valoarea critică a deficitului de fier : <10 mg/ml sau < 10 µg/L• Creşterea feritinei ( >400 µg/L ):
– Supraîncărcarea cu fier din hemocromatoză sau hemosideroză– Administrarea orală sau parenterală de fier– Bolile infamatorii– Bolile acute sau cronice hepatice, implicând alcoolismul– Anemia hemolitică, anemia megaloblastică, talasemia, anemia sideroblastică.
Factori care interferă cu valoarea normală a feritinei:• - administrarea recentă de medicamente radioactive determinând rezultate false,• - contraceptivele orale şi terapia antitiroiană interferă cu testarea,• - hemolizarea sângelui poate determina rezultate crescute,• - creşte cu vârsta,• - mai mare la cei care mânâncă carne roşie, decât la vegetarieni,• - valoarea feritinei nu este o metodă eficace de evaluare a nivelului de fier la persoanele alcoolice afecţiuni
hepatice.

Depozitele de FeDeterminarea sideroblaştilor
• În măduva osoasă, normoblaştii conţin granule de fier ( colorate ) denumite sideroblaşti. Eritrocitele care conţin pete de fier se numesc siderocite. În mod normal sideroblaştii reprezintă aproximativ 33 % din particulele măduvei pe frotiul de măduvă.
• Coloraţia fierului din măduva osoasă este standardul de aur al deficitului de fier: prezenţa fierului exclude deficitul de fier. Dispariţia fierului din măduvă se produce înainte de apariţia modificărilor în sângele periferic, ceea ce apare în anemia prin deficit de fier. Singurii pacienţi care beneficiază de tratamentul cu fier sunt cei la care depozitele medulare de fier sunt scăzute.
• Valori Normale :• Măduva osoasă : 33 % sideroblaşti• Sângele periferic : fără sideroblaşti

Depozitele de FeDeterminarea sideroblaştilor
• se face un frotiu de măduvă osoasă ( poate fi folosit material bioptic de măduvă osoasă), se colorează şi se examinează la microscop prezenţa fierului.
• acest test trebuie făcut şi din sânge periferic pentru detectarea anemiilor sideroblastice.
Implicaţii Clinice :• Scăderea fierul din măduva osoasă :
– deficitul de fier prin toate cauzele de sângerare cronică, malignitate,– policitemia vera– infecţie cronică– boli mieloproliferative
• Creşterea fierul din măduva osoasă în :– hemocromatoza ( primara sau secundara ),– anemia, mai ales talasemia majoră şi minoră, dar şi alte anemii hemolitice,– anemia megaloblastică în recidivă,– infecţii cronice,– insuficienţa pancreatică cronică.
Factorii care interfera : ingestia de fier-dextran va determina valori normale

Anemia feriprivă
• Def - Reprezintă cea mai frecventă formă de anemie. Deficitul de sinteză al hemului apare datorită scăderii cantității de fier total din organism, prin mecanisme diverse: pierderi, deficit de aport, creșterea necesarului de fier.
• Deficitul de fier este indicat de – scăderea sideremiei – creşterea CTLFe– creşterea transferinei– scăderea saturaţiei tramsferinei – Scăderea feritinei– Det. Sideroblaștilor

An. Feriprivă-etape-
• Faza 1 - concentrația hemoglobinei și sideremia sunt normale, dar scad depozitele de fier, pierderea depășind aportul. Feritina scade treptat, timp în care crește absorbția concentrația plasmatică a transferinei.
• Faza 2 – feritina se epuizează, tranferina crește, sideremia scade, deci fierul disponibil pentru eritropoieză scade.La nivel medular va fi afectată etapa de maturație, cu apariția hiperplazie eritroblastice, cu nucleu matur /citoplasmă imatură. Când sideremia scade sub 50%, iar saturația transferinei sub 16% eritropoeza este afectată, la nivel medular scad sideroblaști.
• Faza 3 – apare o anemie normocitară, normocromă.• Faza 4 – anemia devine microcitară și apoi hipocromă.• Faza 5 – deficitul de fier va induce treptat apariția simptomatologiei
nespecifice: astenie, dispnee, paloare, manifestări neuro-musculare, iritabilitate, cefalee, tulburări vaso-motorii și sensibilitate.

An feriprivă
• Tratamentul constă în depistarea şi eliminarea cauzei care produce deficitul de fier,
• la care se asociază administrarea de fier, cu rezultate vizibile din prima lună.
• Un indicator al eficienţei tratamentului este creşterea reticulocitelor.

Anemii sideroblastice
• grup de afecţiuni caracterizate printr-o captare ineficientă a
fierului, având ca efect scăderea sintezei de hemoglobină.
Pot fi
• primare – determinate prin deficit genetic, care se transmit X-
lincat sau autosomal recesiv
• secundare – sunt inhibate enzimele implicate în sinteza
hemului, ca urmare a unor afecţiuni asociate: inflamaţii, cancere,
medicamente( izoniazidă, alcoolism cronic,cloramfenicol),radiaţii.

Anemii sideroblastice
• Anemia sideroblastică primară este o afecţiune rară,
• poate fi piridoxal sensibilă, sau piridoxal insensibilă.
• Forma piridoxal sensibilă
• se caracterizează prin incapacitatea încorporării fierului în protoporfirină, fier
care se depozitează în mitocondrii.
• Adm. de vit. B6 (piridoxină) în doze mari, poate ameliora sinteza.
• Vitamina B6 în forma sa activă de piridoxal fosfat este un cofactor al enzimei
ALA-sintetază, care intervine în sinteza acidului delta-aminolevuric, prima etapă în
sinteza hemului.
• Forma de anemie sideroblastică piridoxan insensibilă
• se caracterizează prin creşterea coproporfirinei din cauza deficitului de
coproporfirinoxidază, care determină trecerea acestuia în protoporfirină, la care se
asociază un deficit funcţional mitocondrial, care împiedică sinteza hemului.

Anemii sideroblastice
• În sângele periferic se evidenţiază o anemie microcitară, hipocromă.
• Absorbţia fierului la nivel intestinal este crescută, creşte sideremia, creşte saturaţia transferinei, scade transferina.
• Diagnosticul se face prin biopsie medulară, unde se evidenţiază o creşterea a sideroblaştilor, cu apariţia unui inel perinuclear caracteristic( sideroblaşti inelari) şi a feritinei.
• Măduva hematogenă este hiperplaziată, datorită stimulării eritropoezei, dar o parte din eritrocite sunt distruse înainte de a fi eliberate în circulaţie.
• Manifestările sunt cele comune anemie la care se asociază semne de supraîncărcare cu fier: hepato şi splenomegalie, piele cu aspect bronzat, tulburări de ritm cardiac, accidente coronarieie prin combinarea fierului cu oxigenul şi formarea de radicali liberi, care oxidează LDL şi lezează endoteliul coronarian, favorizând ateroscleroza.

Anemia din boli cronice• Sideremia – scade• Transferina – scade• Saturația transferinei- scade• CTLFe – scade• Feritina –crește• Sideroblaștii- cresc• Mecanisme - Apar prin activarea sistemului
reticuloendotelial care determină reducerea duratei medii de viaţă prin exacerbarea hemolizei. Fierul eliberat prin hemoliză este sechestrat în sistemul reticuloendotelial, fiind blocat circuitul de reutilizare al fierului după hemoliză. Secundar se asociază o scădere a sintezei de eritropoetină, care nu mai este proporţională cu gradul anemiei.

Teste în anemii hipocrome microcitareTulburări în sinteza lanturilor de hemoglobină
• Talasemiile

Talasemiille
• Talasemiile reprezintă un grup de anemii hemolitice, microcitare congenitale, caracterizate printr-o sinteză defectoasă a hemoglobinei, asociată cu un proces de eritropoieză ineficientă.
• Talasemia este cea mai frecventă anemie hemolitică congenitală, care apare prin scăderea sintezei unuia din lanţurile polipeptidice ale globinei.

Sinteza lanţurilor globinice
• este controlată genetic. • Se cunosc 5 tipuri de subunităţi globinice: α, β, γ, δ, ε, contolate prin
gene diferite.• La adultul normal predomină HbA cu structura α 2 β2 .• Lanţurile β sunt foarte asemănătoare cu lanţurile δ, • genele responsabile fiind situate pe acelaşi cromozom,• lanţurile δ au o viteză de sinteză de 40 de ori mai mică decât
lanţurile β, de aceea HbA2 cu structura α 2δ2 , se găseşte în proporţie de 2,5% din Hb totală.
• Lanţurile ε sunt formate în primul trimestru al vieţii embrionare, • din trimestrul II de sarcină predomină sinteza lanţurilor α şi γ ,
rezultând prin combinare hemoglobina fetală HbF cu structura α 2 γ
2. • După naştere sinteza lanţurilor β creşte rapid după naştere, Hb A
înlocuind treptat HbF, astfel că la 6 luni HbF este sub 2%.

Structura hemoglobinei normale
• favorizează transportul oxigenului,
• subunităţile γ şi β au o structură complementară care se
asociază spontan formând dimeri, care se reunesc printr-un
punct de contact asimetric între lanţul alfa al unui dimer şi
lanţul beta al celuilalt dimer, rezultând o configuraţie completă
tetramerică.
• Acest contact asimetric (α 1 β 2) determină forma normală
sigmoidă a curbei de disociaţie a oxihemoglobinei.

Alfa talasemia• Se caracterizează prin scăderea sintezei lanţurilor
alfa, având un model complex de transmitere genetică. • Forma homozigotă de alfa talasemie cu defect genetic
dublu, este incompatibilă cu viaţa, deoarece lanţurile α nu se sintetizează, rezultând un tetramer de lanţuri γ – hemoglobina Bart, care nu poate transporta oxigenul.
• Deficitul de sinteză a lanţurilor α determină sinteza în exces a tetramerilor de lanţuri β, rezultând Hb H.
• Forma heterozigotă poate fi pentru o singură genă - nu este simptomatică,
• în timp ce defectul genetic dublu induce un tablou clinic similar cu heterozigoţii din beta-talasemie.

Beta talasemia
• Se caracterizează prin scăderea sintezei lanţurilor β.
• Heterozigoţii sunt purtători şi au o anemie microcitară medie,
asimptomatică – talasemia minoră.
• La homozigoţi apare β- talasemia majoră cunoscută şi sub numele de
anemia Cooley.
• Pacienţii prezintă anemie severă, icter, splenomegalie, sideroză hepatică,
hiperplazie măduvei hematogene şi supraîncărcarea cu fier, datorită
transfuziilor repetate.
• Hiperactivitatea măduvei hematogene produce îngroşarea oaselor
craniene , a eminenţelor malare, la oasele lungi producând fracturi
patologice.

Siclemia – boala celulelor în seceră
• este un termen pentru un grup de boli ereditare ale sangelui.
• Anemia celulelor in secera este o cauza de Hb anormala,
• Presupune o alterare a secvenței de aac. la nivelul unui lanț de globină, fără o
reducere în rata de sinteză a lanțului anormal. Rezultă mai multe tipuri de Hb : S, C,
E, D, etc.
• În siclemie acidul glutamic din poziția 6 a lanțului beta normal, este înlocuit cu
o moleculă de valină. Hb S redusă este de 50 de ori mai puțin solubilă decât Hb A
redusă.
• În hipoxie HbS deoxigenată (redusă), formează polimeri, apoi fibre lungi care
deformează hematia într-o formă tipică de “ seceră” – polimerii de Hb se așază pe
axul lung al hematiei, iar membrana se pliază pe acest conținut rigid. Pt.
heterozigoți fenomenul de siclemie apare la pres. scăzute ale O2.
• Afecțiunea este frecvent prezentă în Africa tropicală.

Siclemia – boala celulelor în seceră
• Inițial există cicluri de siclizare, hematiile revin la forma inițială.
• Ulterior deformarea este ireversibilă. Atât hematiile siclizate cât și cele nesiclizate sunt mai
puțin plastice.
• Ele produc fenomenul de încetinire a circulației cu obstrucția vaselor mici, cu infarct tisular
consecutiv.
• Stare homozigotă a bolii cu HbS este asociata cu morbiditate considerabila si mortalitate.
• Starea heterozigotica prezinta mortalitate mica.
Clinic – creează tabloul de anemie cronică – 6 –9 g /dl
- cutanat – ulcere de gambă – infirmizante
- crize dureroase articulare, sdr. toracic, afectarea SNC( AVC), oasele (femur, humerus),
- ap. genital – priapismul –impoteță sexuală
- autosplenectomie – inf. severe.
DG.- ELFO Hb - model homozigotic ( S/S ): HbS, 80% - 100%; HbF, restul cea mai ramas; HbA1 0%
( absenta )

Teste -Electroforeza hemoglobinei• Hemoglobina normală şi anormală poate fi detectată prin electroforeză, care constă în
hemoliza hematiilor, urmată de migrarea materialului hematic pe benzile standard în funcţie
de tipul de Hb cunoscute.
• Cele mai comune forme de Hb adultă normală sunt HbA1, HbA2 şi HbF ( Hb fetala ).
• Cele mai cunoscute hemoglobine anormale ( hemoglobinopatii ) sunt HbS ( responsabila de
anemia cu celule în seceră ) şi HbC ( rezultă în anemia hemolitică uşoară ).
Atenţie!!! Rezultatele trebuie să fie interpretate în funţie de transfuzile de sânge efectuate în
lunile precedente testarii.
Valori Normale :
• HbA1: 96.5 % - 98.5 % sau 0.96 – 0.98 fracţie de masa,
• HbA2 : 1.5% - 3.5 % sau 0.015 – 0.035 fracţie de masa,
• HbF ; 0% - 1% sau 0 0.01 fracţie de masa.

Hemoglobina Fetală -(hemoglobina F; hemoglobina alcalin-rezistenta )
• HbF este o Hb normală a fetusului şi a copilului; reprezintă 50% până la 90 % din Hb nou-născutului, restul reprezentând HbA1 şi HbA2.
• În condiţii normale, fabricarea de HbF este înlocuită de fabricarea de tipuri adulte de Hb în timpul primului an de viaţă.
• Dar daca HbF persista şi constituie mai mult de 5% din Hb după vârsta de 6 luni, o anomalie trebuie suspectată.
• Determinarea HbF este utilizată în evaluarea talasemiei, anemii hemolitice, persistenţa ereditară a Hb fetale şi alte hemoglobinopatii.
Valori Normale:• Adulţi : 0% - 2% sau 0 – 0.02 fracţie de masă HbF• Nou-născuţi : 60% - 90% sau 0.60 – 0.90 masa de fracţie HbF• La vârsta de 6 luni : 2% sau 0.02 masă de fracţie HbF

Teste speciale în an. macrocitară , normocromă
• Dozarea vit. B12
• Dozarea ac. folic

Dozarea vit. B12• Vitamina B12 este o substanță complexă, a cărei absorbție se face în
prezența factorului intrinsec la nivelul ileonului terminal. Se obține prin aport de proteine animale.
Valori normale:• Cantitatea de vitamina B12 - adult = 200-835 pg/ml - n.n.= 160-1300pg/ml• Capacitatea de legare a vitaminei B12 - adult = 600-1400 pg/mlInterpretare clinicăScăderea vitaminei B12 apare în:• aport deficitar• sindromul de malabsorbție – este cel mai frecvent mecanism, care se
produce prin: deficit de producție a factorului intrinsec( gastrectomii, anemie Birmer), inhibiția factorului intrinsec, afectarea ileonului terminal (rezecții, boli congenitale, boala Crohn’s), inhibiția competitivă a absorbției prin infestare cu Diphyllobothrium latum.

Dozarea vit. B12Creşterea concentrației de vitamină B12:• IRC, leucemie cronică granulocitară• boli hepatice• policitemia vera• BPOC, diabetTestul Schilling• Scop – evidențiază tulburarea de absorbție a vitaminei B12, putând fi între variatele cauze de malabsorbție.Etape:• diagnosticul de absorbție scăzută/normală• se administrează vitamina B12 marcată cu Co57 p.o. (oral), iar pentru a evita utilizarea vitaminei B12 în
țesuturi, se injectează 1 mg de vitamină B12 i.m. Ne interesează să urmărim cantitatea de vitamină B12 care trece din intestin, în sânge și apoi în urină.
• Se colectează urina în primele 24 h, și se calculează radioactivitatea ei.v.n.= 10 – 40% din cantitatea de vitamină B12 marcată cu Co57 care s-a administrat se dozează în urină.• < 7- 8% = absorbție anormală, este obligatorie a 2-a etapa. Dacă pacientul are IRC se prelungește timpul de
recoltare al urinei!!! • Se va administra după 2 săptămâni:• vitamina B12 marcată cu Co 58• vitamina B12 marcată cu Co 57 + factor intrinsec• Se recoltează urina și se determină radioactivitatea ei, pt. fiecare în parte.Interpretare: vitamina B 12 se elimină urinar =deficit de factorul intrinsec sau dacă vitamina B12 nu se elimină prin urină sau scaun = deficit de absorbție ileală.

Dozarea ac. folic• Acidul folic este o vitamină care se găsește în abundență în legumele și fructele proaspete,
fiind rapid distrusă de temperaturile înalte. Necesarul zilnic este de 50 g/zi ( față de Fe(10000g/zi). Rezervele organismului sunt suficiente pentru aproximativ 4 luni.
• Se absoarbe la nivelul jejunului proximal. El este un metabolit inactiv care este redus la forma activă de acid tetrahidrofolic de către dihidrofolat reductaza ( este inhibată de medicamentele antineoplazice – metrotrexat). Scăderea acidului folic are manifestări asemănătoare cu cele din carența de vitamină B12 , dar lipsesc semnele neurologice.
Valori normale • Adulți 2-20 ng/ml• Copii 5-21 ng/ml• n.n.14-51 ng/mlScăderea folaţilor apare:• -anemii hemolitice si megaloblastice• - consum crescut - la alcoolici, copii cu aport exclusiv de lactate, femei însărcinate, • - sindromul de malabsorbție,• - deficit de absorbție - medicamente care scad absorbția folaților ( fenitoin, fenobarbital,
izoniazidă, cicloserina).Creşterea apare la:• - vegetarieni,• - anemie Birmer.

Anemia Birmer sau pernicioasă
• este cea mai frecventă anemie prin careţa de vitamina B12. • Apare la persoanele peste 30 de ani Mecanism patogenetic • Apare prin scăderea absorbției vitaminei B12 din cauza lipsei factorului intrinsec. • Deficitul poate fi congenital sau dobândit prin atrofia mucoasei gastrice,
gastrectomie. • Aspectul megaloblastic este rezultatul perturbării balanței între cantitatea
neceară de cofactori pentru sinteza de AND și cantitatea de AND sintetizată. Cei mai importanți 2 cofactori sunt :vitamina B12 și ac folic. Ei sunt implicați în sinteza timinei. Lipsa lor duce la sinteza de timidina, care va induce un defect de sinteză al AND, deci un deficit de diviziune celulară.
• Macrocitoza presupune un asincronism nucleu citoplasmă, secundar deficitului de vitamina B12 și acid folic, reprezentat prin creșterea cantității de citoplasmă în comparație cu cantitatea de acizi nucleici, fără ca aceștia să se mai dividă.
• Macrocitoza poate să apară și în – hipoglicemii repetate, alcoolici.

Anemia Birmer sau pernicioasă
• Concomitent înafara măduvei hematogene mai este afectată şi teaca de mielină –determinând tulburări neurologice prin demielinizarea măduvei spinării şi cerebelului, şi afectarea mucoasei tubului digestiv care determină achilie gastrică.
• Cu cât deficitul este mai sever, cu atât eritropoeza este ineficientă. Dacă măduva este hiperplazică, megaloblaştii sunt prezenţi pe aproape toate liniile celulare.
• Pentru diferenţiere se face testul Schilling şi se determină în sânge acidul folic şi ciancobalamina.
• Fe poate fi crescut uşor dacă există hemoliză şi poate să devină deficitar dacă începem terapia cu vitamină B12, de aceea se recomandă asocierea la tratamentul cu vitamina B12 şi acid folic, a fierului.

Teste speciale în anemia normocitară normocromă
• În aceste anemii indicii eritrocitari sunt normali în condițiile în care numărul total de eritrocite este scăzut.
• Sunt forme rare de anemii, cu mecanisme multiple de producere: – tulburări de formare,– tulburări de maturare a precursorilor hematopoetici la
nivel medular, – deficite enzimatice– anemii posthemoragice.

Anemia Hemolitică
• Anemiile hemolitice sunt afecțiuni congenitale sau dobândite, care au drept caracteristică comună scurtarea duratei de viață a hematiilor.
• Hemoliza se poate produce– intravascular (rar, liza vasculară cu eliberarea hemoglobinei)
• sau extravascular ( prin intermediul sistemului monocit - macrofag din ficat, splină, măduvă osoasă).
Anemiile hemolitice pot fi împărțite în:• anemii hemolitice prin defect de membrană eritrocitară• anemii hemolitice prin defecte enzimatice• anemii hemolitice prin defect de sinteză a hemoglobinei -

Teste speciale în anemii hemolitice prin defect enzimatic la nivelul membranei eritrocitară
• Dozarea piruvatkinazei

Dozarea piruvatkinazei
• Deficitul de piruvatkinază este o tulburare transmisă genetic caracterizată prin scăderea concentrației de adenozintrifosfat de la nivelul eritrocitelor, cel mai frecvent defect enzimatic ereditar al enzimelor din calea de glicoliză anaerobă.
• Lipsa enzimei va genera un deficit de ATP, necesar funcționării pompei ionice de Na-K, alterand gradienții ionici transmembranari.
• Deficitul genetic se transmite autosomal recesiv, determină rigidizarea membranei,care va induce o anemie hemolitică cronică nonsferocitară.
Valoare normală: 2,8 – 8,8 U/g Hg. • Pentru a converti in U/ml: U/g Hg X 0,34 = U / ml • Procedura: 5 ml de sange pe EDTA se congeleaza imediat.

Scăderea piruvatkinazei semnifică
• deficit congenital transmis autosomal recesiv, bine tolerat pentru că scade și 2,3 difosfoglicerat.
• deficit dobândit apare în : tulburări mielodisplazice, leucemie acută, anemie.
• În deficitul congenital de piruvatkinaza hemoliza se manifestă în timpul sarcinii sau când se folosesc contraceptive orale.
• În anemiile severe se impune efectuarea splenectomiei.• Unele medicamente interferează cu funcția
hemoglobinelor. De exemplu : sulfamide, antipiretice, analgezice, doze mari de vitamina K, nitrofurantoina.

Teste speciale în anemii hemolitice prin defect de membrană eritrocitară –sferocitoza ereditară
• Teste de autohemoliză

Sferocitoza ereditară
• Boala se caracterizează prin apariția unui defect de membrană eritrocitară, datorită reducerii conținutului lipidic care va determina scăderea suprafeței membranei eritrocitare, aceasta devenind sferică.
• Permeabilitatea membranei se modifică, K+ iese din celulă și Na+ intră, modificând gradientul ionic al celulei printr-un consum crescut de ATP, necesar pentru activarea pompei Na-K.
• Consumul crescut de ATP va determina o creștere a consumului de glucoză. • Scăderea aportului de glucoză prin hipoglicemie sau a glicolizei, va
determina acumularea sodiului în eritrocite.Sodiul atrage apa și determină creșterea volumului eritrocitelor, cu ruperea membranei eritrocitare și hemoliză.
• Sferocitele având un volum mic se rup repede prin creșterea volumului eritrocitar. Așa cum știm sediul unde hemoliza este cea mai mare este în splină, unde la nivelul pulpei splenice concentrația glucozei este scăzută, iar pH este acid, limitând glicoliza.
• Sferocitele devin rigide, nu pot străbate fenestrațiile de la nivelul pulpei roșii splenice, unde sunt captate și distruse de macrofage.

Determinarea fragilității eritrocitelor sau autohemoliza
• hematiile au formă de disc biconcav.
• Această formă însă este observată la hematiile examinate la microscop, deoarece
hematiile circulante sunt supuse unui stres circulator care le deformează.
• Cea mai mare deformare se produce la nivelul splinei când forma de disc biconcav se
pierde. Există situaţii în care capacitatea de deformare a hematiei se pierde, de exemplu:
sferocifoza erediatară.
• Sferocitele apărute prin orice modalitate ( sferocitoza ereditara, toxice ) sunt susceptibile
la procesul de hemoliză în soluţii saline (hipertone), mai intens decât hematiile normale.
• În general, celulele sferice ( rotunde ) au o fragilitate osmotică crescută, iar celulele cu
creşterea raportului suprafaţă / volum ( celule subţiri, turtite şi hipocrome ) au o fragilitate
osmotică scăzută.
• În sferocitoza ereditară testul de fragilitate osmotică poate fi iniţial normal, dar incubarea
la 37 º C , 24 h determina pozitivarea testului.

Valori normale
- hemoliza imediată – hemoliza începe la concentrația de 0,5 %
NaCl, iar hemoliza este completă la concentrația de 0,3 % NaCl.
- după 24 h de incubare - hemoliza începe la concentrația de
0,7 % NaCl, iar hemoliza este completă la concentrația de
0,4 % NaCl.
Procedura: 7 ml de sânge pe substrat anticoagulant ( heparina ).
Se supun eritrocitele la diluții variate de NaCl. Se citește hemoliza
la spectofotometru ( măsoară densitatea optică ).

Implicații clinicecreșterea fragilității osmotice: • anemie hemolitică ( autoimuna ),• sferocitoza ereditară,• boala hemolitică a nou-nascutului,• malaria,• deficit sever de piruvatkinază,scăderea fragilității osmotice: - anemie prin deficit de fier,• anemie microcitară hipocromă,• talasemia,• postsplenectomie,• boala hepatică,• reticulocitoza,• hemoglobinopatii ( HbC, HbS ) .

Teste speciale în anemii hemolitice prin deficit de enzime intracitoplasmatice
• Dozarea glucozo-6-fosfat –dehidrogenazei
• Determinarea corpilor Heintz
• Deficitul de 2,3 difosfoglicerol

Glucozo-6-fosfat-dehidrogenaza
• Este o tulburare cu transmisie genetica X – lincata. Apare in grupuri etnice, mai frecvent la bărbaţii afro-americani, 11 % din barbatii homozigoti, 20 % din femeile heterozigote. Este rară în Ro.
• Deficiența afectează o enzimă din șuntul pentozofosfaților, determină scăderea nivelului de ( NADPH și GSH) produși oxidanți în exces, care produc o sensibilitate mărită a hematiei la acțiunea unor agenți reducători (penicilină, vit. K, vit. C, naftalina, coloranți, infecții, alimente-fasole).
• Eritrocitele acestor bolnavi nu pot menține un nivel normal al glutationului redus, în consecință grupările SH din hemoglobină se oxidează și precipită formând corpusculi Heintz, sub influența unor substanțe medicamentoase, sau a unor infecții virale se produce la 24- 48 ore o hemoliză brutală.
• Corpi Heinz vor fi extrași la nivelul splinei de către macrofagele splenice, eritrocitele rezultate vor fi bine colorate dar vor prezenta la periferie zone “amputate”. Ulterior, hematiile vor fi lizate în splină.
• Exista o forma de deficit de G6PD indolenta in India, Grecia, Turcia, Africa, Irac, Kârdistan ( Asia de Sud-Est ).

Glucozo-6-fosfat-dehidrogenaza
• Valori normale G6PD:
adult : 8,6 – 18,6 U/g Hg
copii : 6,4 – 15,6 U/g Hg
nou- nascut : valori cu 50% mai mari fata de adult.
G6PD(U/g) X 0,34 = U/ml
•Procedura: 5 ml de sange + EDTA / heparina
Interpretarea rezultate
Scăderea valorilor
•– deficit de G6PD ( in episod hemolitic dupa consum de fasole, medicamente )
• - anemie congenitala nonsferocitara
•- boala hemolitica nonimunologica a nou-nascutului
Creşterea valorilor
•– anemie megaloblastica netratata
•- purpura trombocitopenica
• - hipertiroidism
• - hepatită virală.
!!! Reticulocitoza crescută poate determina valori fals crescute ale G6PD. G6PD poate avea valori fals normale intre 6 si 8
saptamani de viaţă, după un episod hemolitic, mai ales la sugari. Dupa episoade de hemoliza testul trebuie refacut.

Corpii Heintz
• Corpusculilor Heinz sunt incluziuni insolubile intracelulare ale hemoglobinei, fixate pe membrana eritrocitului. Ei sunt intalniti doar in deficitul de G6PD imediat după hemoliza şi la pacienţii cu forme de Hg instabilă.
• Corpusculii Heinz apar probabil prin precipitarea hemoglobinei instabile ( oxidarea Hb denaturate); sunt eliminaţi in splină, astfel că după splenectomie ei cresc în sângele periferic fiind găsiţi la 50% din eritrocite.
Interpretare - În mod normal ei nu se găsesc la persoane sănătoase.• Tehnica : - sange venos + heparina / EDTA• - amestec de celule + sustrat colorant ex. MI. Substratul
coloreaza corpusculii in albastru ( pal deschis ) in contrast cu roşu închis al ARN reticulocitar

Corpii Heintz
Cresc în : • deficit de G6PD dupa hemoliză• anemie hemolitica congenitala cu corpusculi Heinz
• variante de hemoglobina instabila ( Zurich Hb,
Phily Hb )• β- talasemie hemozigota• Se găsesc în mod normal la persoane sănătoase• după intoxicaţie cu unele medicamente – cloruri,
fenilhidrazina• la nou-nascuti sau pacienti splenectomizati.

Deficit de 2,3 DPG ( 2,3 difosfoglicerol )• 2,3 difosfoglicerol asigură transportul de oxigen la nivelul eritrocitului.
- creşterea 2,3 DPG apare în hipoxemie sau anemie- scăderea 2,3 DPG apare în acidoza.
• În mod normal nivelul 2,3 DPG este scăzut şi la nou-născuţi şi foarte scăzut la prematuri.Valori normale :adulti: 10,4 – 14,2 mmol/g HgInterpretare:Creşterea 2,3 DPG- emfizem, fibroza chistică• boala cardiaca cianogena• boala pulmonara vasculara• anemie celulara – siclemie, anemie cu deficit de fier• deficit de piruvatkinaza• hipotiroidism• IRC• ciroza• creşterea altitudinii• exercitii fiziceScăderea 2,3 DPG – policitemia vera• sindrom de detresa respiratorie• deficit de 2,3 DPG • deficit de hexokinaza! Dacă la transfuzii se administreaza sânge cu deficit de 2,3 DPG, Hb acestuia nu va elibera O2 când este necesar.

Anemia hemolitică autoimună
• Este o anemie dobândită care rezultă din distrugerea prematură
a eritrocitelor, cauzată de prezența anticorpilor și /sau a
complementului pe suprafața hematiei.
• Anticorpii prezenți sunt de trei tipuri:
- Ac reactivi la temperatura corpului – autoanticorpi la cald de tip Ig G,
- Ac reactivi la rece - autoanticorpi la recede tip IgM,
- Ac formați în urma imunizării prin transfuzii repetate sau sarcină,
orientați împortiva eritrocitelor transfuzate.
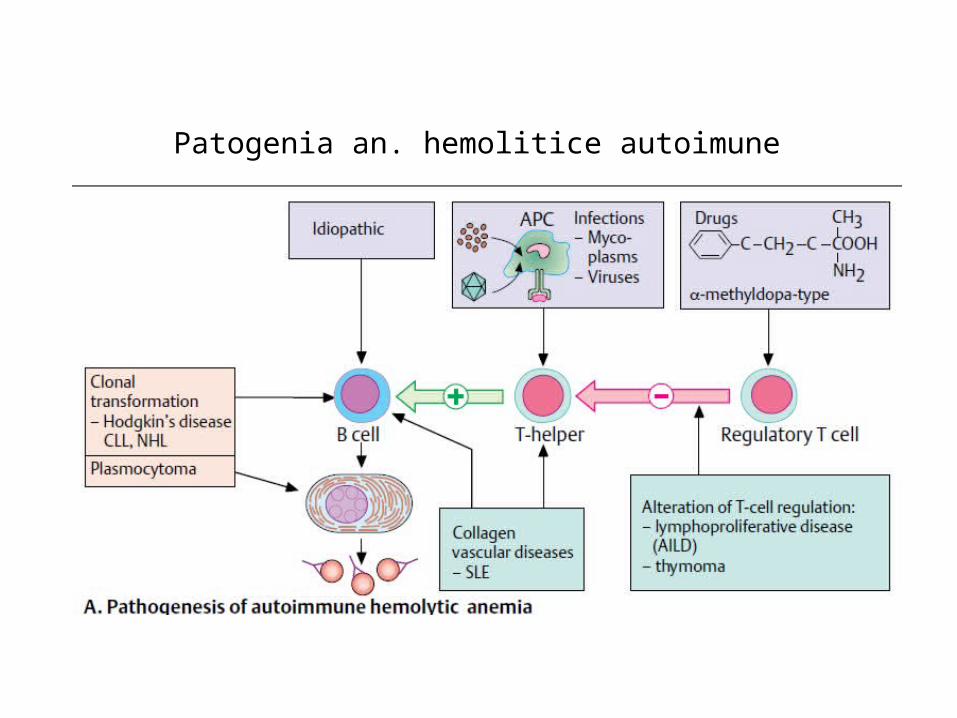
Patogenia an. hemolitice autoimune

Anemia hemolitică autoimună
• Hematiile învelite în IgG interacționează cu receptorii Fc ai macrofagelor, fiind fagocitate.
Dacă fagocitoza este parțială eritrocitele restante vor circula sub formă de sferocite.
• Când intervine complementul hematiile sunt învelite în fracțiunea C3, care se va activa și va
reacționa cu receptorii macrofagici, fiind complet fagocitate
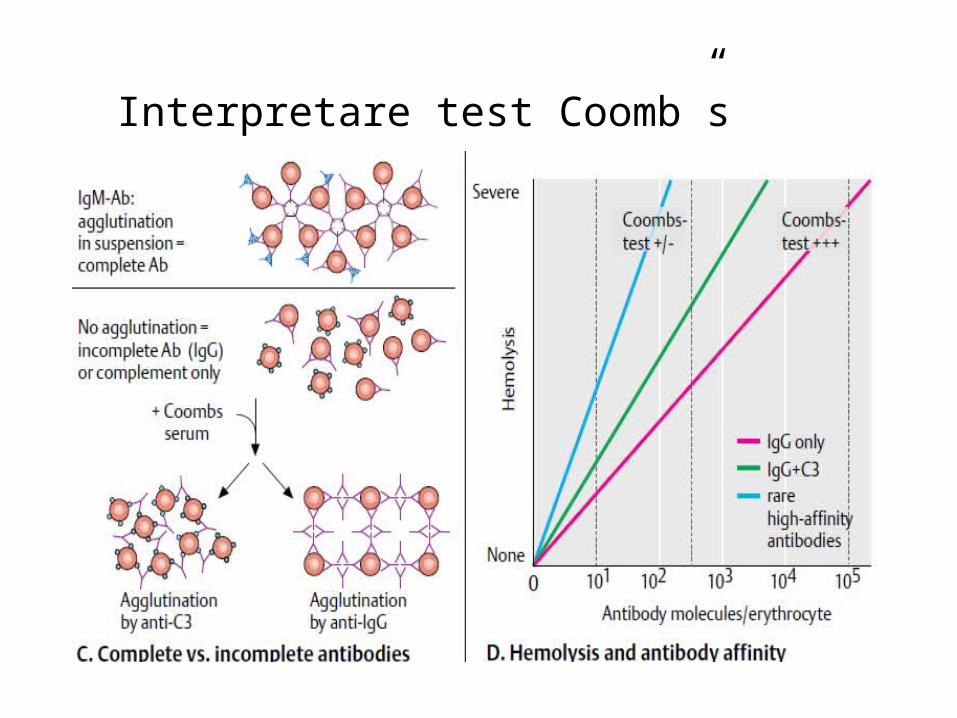
În anemia hemolitică cu Ac la cald:
• frecventă la femei 30-60 ani, hemoliză extravasculară,
• Ac tip IgG se fixează direct pe suprafața membranei eritrocitare, care sunt captate apoi de
macrofagele splenice suferind o transformare sferocitară.
• Complexul Ac la cald -hematie este inițiat de IgG sau fracțiunea C3, și este foarte grav la bolnavii
care prezintă ambele componente pe membrana eritrocitară,
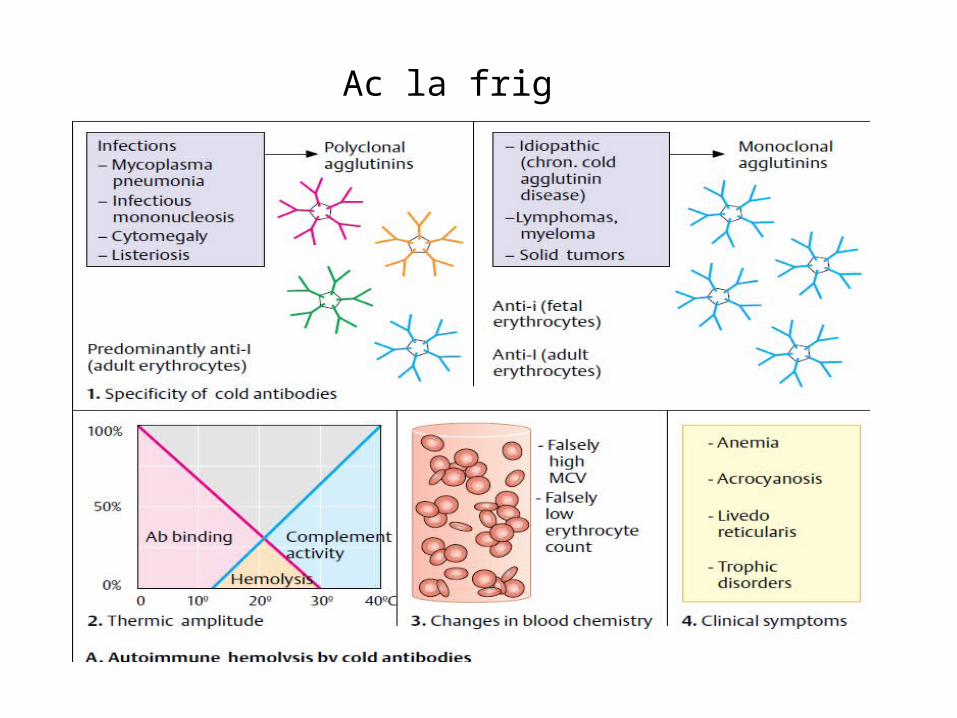
• Test Coombs direct este pozitiv pentru IgG și complement.În anemia hemolitică cu Ac la rece • frecventă la bărbați, 50-80 ani, Hemoliza este intra sau extravasculară,• autoanticorpii la rece sunt de tip IgMsau IgG, producând hemoliză prin fixearea componentei
complementul C3 la suprafața eritrocitelor,• Test Coombs este negativ pentru Ig și pozitiv pentru complement.
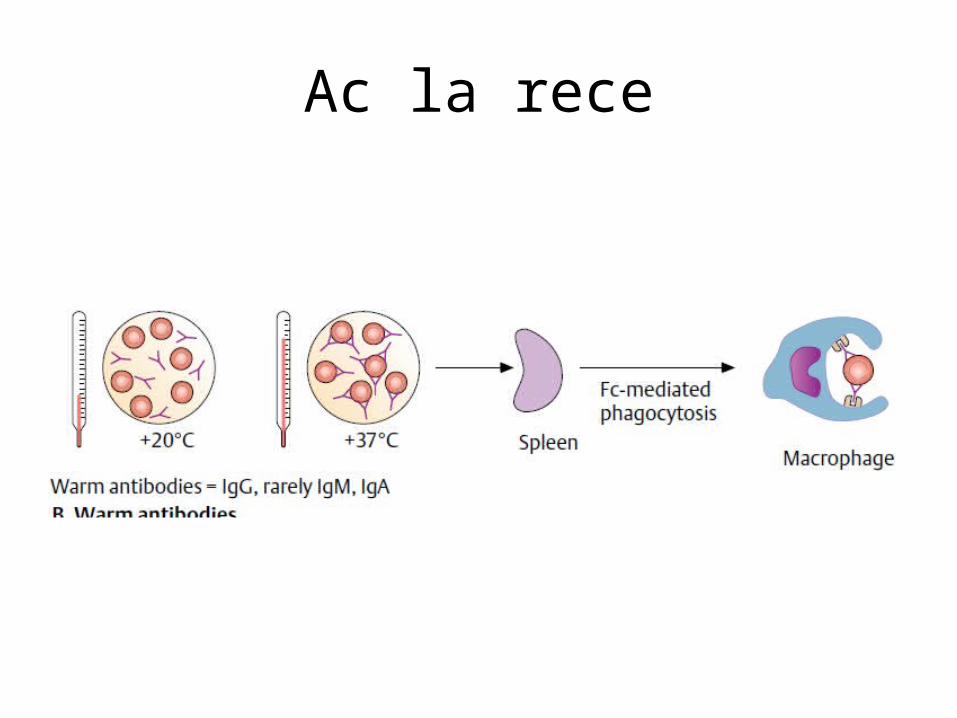
Ac la rece

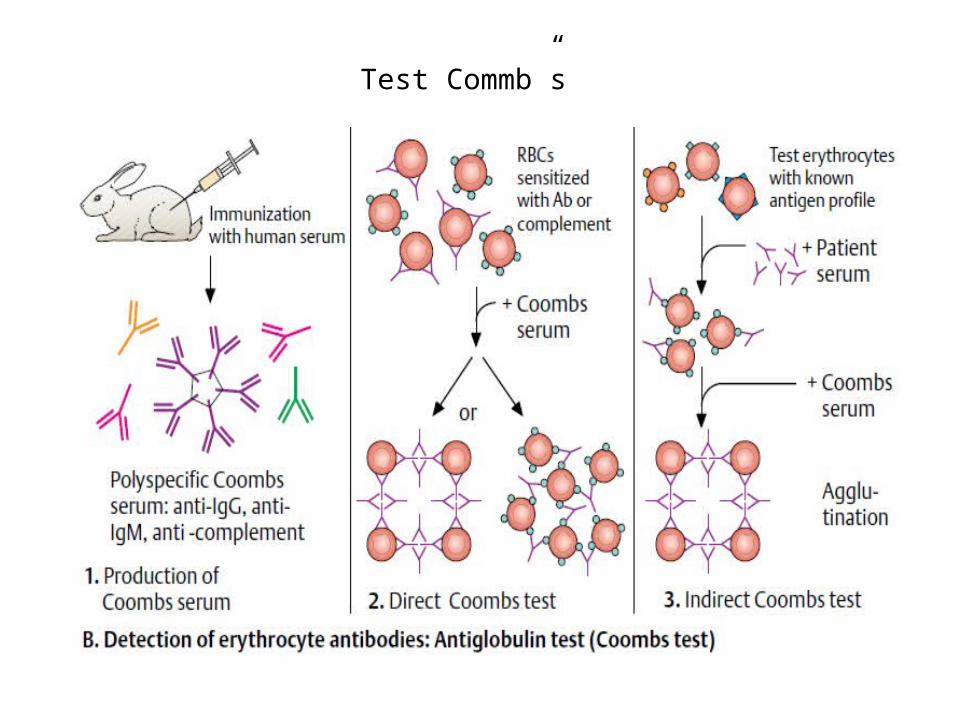
Testul Coombs
Se utilizează în diagnosticul anemiilor hemolitice autoimune:
• testul Coombs direct evidențiază anticorpii antieritrocitari fixați pe eritrocite
• testul Coombs indirect evidențiază anticorpii antieritrocitari liberi din ser.
Interpretare
în mod normal ambele teste Coombs direct și indirect sunt negative
• Reacții pozitive (1+ - 4+ ) pot să apară în următoarele situații:
- reacție posttransfuzie,
- anemie hemolitică autoimună( în 75% din cazuri),
- tratamente cu penicilină, insulină, α-metildopa, cefalotină, alte medicamente,
- boala hemolitică a nou-născutului,
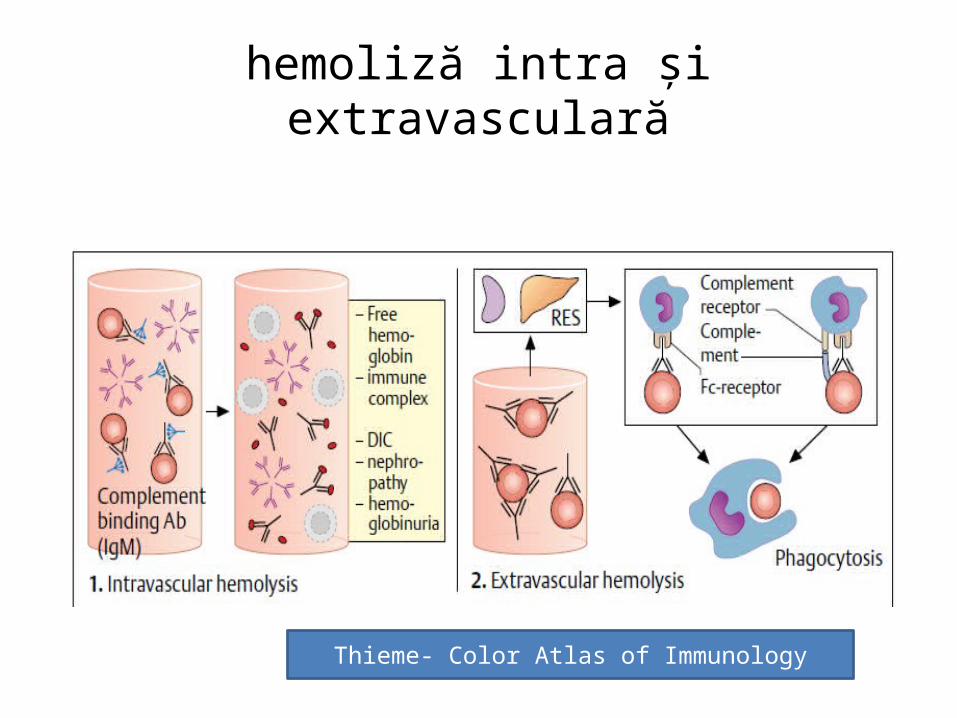
hemoliză intra și extravasculară
Thieme- Color Atlas of Immunology
Test Commb”s
Interpretare test Coomb”s
clinic
Ac la frig
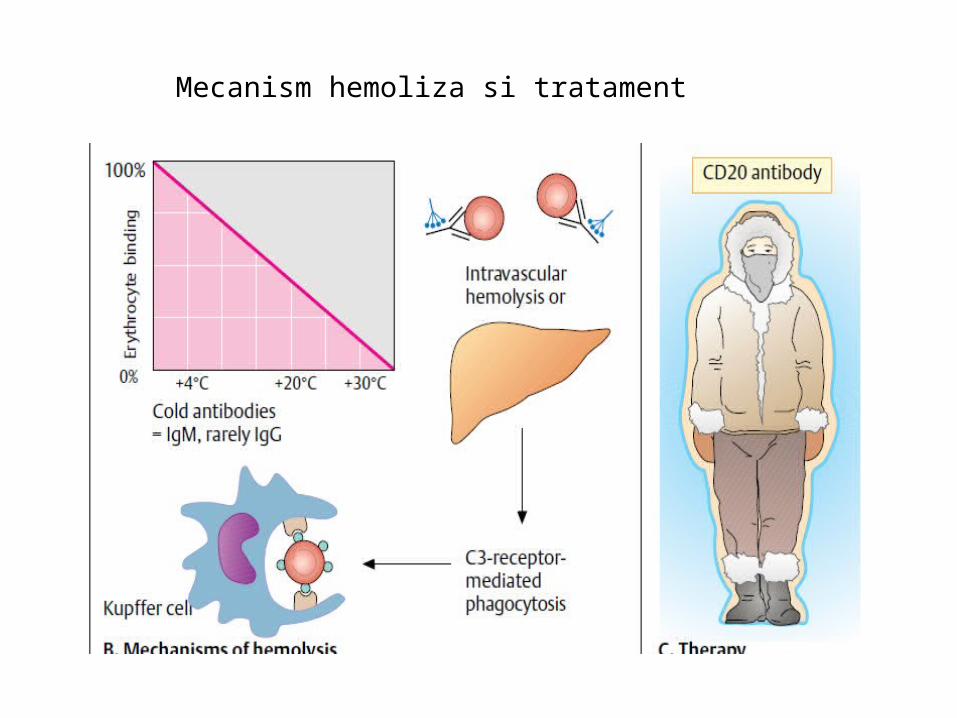
Mecanism hemoliza si tratament

Anemia posthemoragică
• Forme:– acută ( peste 750 ml) – normocrome, normocitare, regenerative– cronică - normocrome, normocitare, regenerative
Aceste anemii pot imita o anemie feririvă, cu un deficit de acid folic• În forma acută, după o hemoragie masivă apare fenomenul de
hemoconcentrație ( Hb este inițial in limite normale, datorită mobilizării sângelui din depozite,
• la aprox. 1 oră apare trombocitoza (800.000),• la 6 ore leucocitoză (20.000) cu neutrofilie, cu scăderea lor progresivă=
reacție de stress.• În următoarele 12-48 h se produce hemodiluția progresivă,• cu creșterea secundară a reticulocitelor, după 3 - 4 zile.• Criza reticulocitară (funcția majoră a măduvei) apare la 7- 10 zile. • Monoblaștii apar periferic după hemoragiii severe.

An aplastică
• Apare ca urmare a unei hipoplazii severe a seriei eritrocitare, mieloide și trombocitare.
Măduva hematogenă este substituită cu țesut adipos.
• Hemoleucograma prezină pancitopenie ( inhibarea tuturor seriilor hematoformatoare),
anemie gravă, refractară la tratament reticulocitele – mult scăzute, sub 1%. Diagnosticul este
susținut de biopsia medulară.
Cauze
• idiopatică, aproape 50% din cazuri,
• intoxicația – hidrocarburi aromatice, insecticide organo-fosforice,
• medicamente – cloramfenicol,
• radiații ionizante,
• inf. virale –HIV, VEB,
• boli de colagen –LES, PAR,
• ereditară – Sindromul Fanconii –boală genetică cu transm autozomal recesivă, caracterizată de
deficit de diferențiere pe toate liniile, care poarta numele de pancitopenie.
Anemia aplastică

Porfiriile
Porfiriile sunt un grup de boli rare caracterizate prin defect de sinteză al hemului, cu
consecințe în acumularea de porfirine sau precursori ai acestora care vor determina
apariția de manifestări neurologice și cutanate.
În porfirie defectul de sinteză este unul enzimatic, în timp ce porfirinuriile presupun
eliminarea crescută și izolată de coproporfirine și/sau uroporfirine - eliminarea
crescută apare în boli hepatice, hepatom, boală Hodkin, afecțiuni neurologice
• Clinic
- Creșterea precursorilor porfirinici - protoporfirinele – determină apariția de
manifestări neuropsihice și crize de dureri abdominale – acestea apar prin mecanism
neuropat, prin inhibarea ATP-azei Na-K dependentă, de către acești precursori care au
o structură asemănătoare acidului-γ-amino-butiric. Se produc la nivel sistemului
nervos central vacuolizări , demielinizări și pareze.

Porfiriile
- Deficitul enzimatic, însoțit sau nu de creșterea precursorilor, determină apariția de
manifestări cutanate. Razele ultraviolete activează porfirinele din vasele de sânge, care
transformă oxigenul în radicali liberi foarte nocivi, pentru țesuturi, prin liza organitelor
celulare și eliberarea de enzime, care produc eritem și leziuni veziculo-buloase. În jurul
capilarelor se acumulează și material amorf.
- Dozarea porfirinelor se face la persoane cu anemie și simptome sugestive: manifestări
neurologice inexplicabile, dureri abdominale inexplicabile, vezicule cutanate, istoric
Precauții pentru persoanele cu porfirie:
• nu este indicată expunerea la familiar.soare la cei cu manifestări cutanate,
• la cei cu manifestări neurologice – atacurile pot fi precipitate de : infecții, ciclul
menstrual, oboseală, medicamente ( barbiturice, estrogeni, etanol, meprobamat,
sulfonamide),