anexa i rezumatul caracteristicilor...
TRANSCRIPT

1
ANEXA I
REZUMATUL CARACTERISTICILOR PRODUSULUI

2
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI Privigen 100 mg/ml soluţie perfuzabilă 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ Imunoglobulină umană normală (Ig IV) Un ml conţine: proteină plasmatică umană…………………………………………………………………………...100 mg (dintre care minim 98% este IgG) Un flacon de 50 ml conţine: 5 g Un flacon de 100 ml conţine: 10 g Un flacon de 200 ml conţine: 20 g Împărţirea subclaselor de IgG (valori medii): IgG1 ............. 67,8% IgG2 ............. 28,7% IgG3 ............. 2,3% IgG4 ............. 1,2% Conţinutul maxim de IgA este de 0,025 mg/ml (media este de 0,0027 mg/ml). Pentru lista tuturor excipienţilor, vezi pct. 6.1. 3. FORMA FARMACEUTICĂ Soluţie perfuzabilă. Soluţia este limpede sau uşor opalescentă şi incoloră până la galben pal. Privigen este o soluţie izotonă, cu o osmolaritate de 320 mOsmol/kg. 4. DATE CLINICE 4.1 Indicaţii terapeutice Terapie de substituţie în • Sindroame de imunodeficienţă primară (IDP), ca de exemplu:
– agamaglobulinemie şi hipogamaglobulinemie congenitale – imunodeficienţe comune variabile – imunodeficienţe combinate severe – sindrom Wiskott Aldrich
• Mielom sau leucemie limfocitară cronică cu hipogamaglobulinemie secundară severă şi infecţii
recurente. • Copii cu SIDA congenitală şi cu infecţii recurente. Imunomodulare • Purpură trombocitopenică imună (PTI), la copii sau adulţi cu risc crescut de sângerare sau
anterior unei intervenţii chirurgicale, pentru a corecta numărul de trombocite. • Sindrom Guillain Barré • Boală Kawasaki

3
Transplant alogen de măduvă osoasă 4.2 Doze şi mod de administrare Doze Doza şi schema de dozaj se stabilesc în funcţie de indicaţii. În terapia de substituţie, este posibil să fie nevoie ca doza administrată să fie ajustată individual în funcţie de răspunsul farmacocinetic şi de evoluţia clinică. Prezentăm câteva scheme terapeutice ca recomandări. Terapie de substituţie în sindroamele de imunodeficienţă primară Schema de dozaj utilizată trebuie să determine obţinerea unei valori minime a IgG (măsurate înainte de administrarea următoarei perfuzii) de cel puţin 4-6 g/l. Pentru echilibrare sunt necesare trei până la şase luni de la iniţierea terapiei. Doza iniţială recomandată este de 0,4 – 0,8 g/kg, continuând cu o doză de cel puţin 0,2 g/kg la fiecare trei săptămâni. Doza necesară pentru a atinge valoarea minimă de 6 g/l este de aproximativ 0,2-0,8 g/kg şi lună. După ce s-a atins starea de echilibru, intervalul dintre administrări variază între două şi patru săptămâni. Pentru a ajusta dozele şi intervalul dintre administrări trebuie determinate valorile minime.. Terapie de substituţie în mielom sau în leucemie limfocitară cronică cu hipogamaglobulinemie secundară severă şi infecţii recurente; terapie de substituţie la copii cu SIDA şi infecţii recurente Doza recomandată este de 0,2-0,4 g/kg la fiecare trei - patru săptămâni. Purpură trombocitopenică imună Pentru tratamentul unui episod acut se recomandă o doză de 0,8 - 1 g/kg în prima zi, care poate fi repetată o dată la trei zile sau 0,4 g/kg zilnic, timp de două până la cinci zile. Tratamentul poate fi repetat în caz de recidivă. Sindrom Guillain Barré 0,4 g/kg şi zi timp de 3 până la 7 zile. Experienţa utilizării la copii este limitată. Boala Kawasaki 1,6-2,0 g/kg trebuie administrate în doze fracţionate într-un interval de două până la cinci zile sau 2,0 g/kg ca doză unică. Pacienţii trebuie să primească tratament concomitent cu acid acetilsalicilic. Transplant alogen de măduvă osoasă Tratamentul cu imunoglobuline umane normale poate fi utilizat ca parte a tratamentului de pregătire şi după transplant. Pentru tratamentul infecţiilor şi profilaxia bolii de tip grefă contra gazdă, dozajul trebuie ajustat individual. În mod normal, doza iniţială este de 0,5 g/kg şi săptămână, începând cu şapte zile înainte de transplant şi continuând până la trei luni post-transplant. Dacă producţia de anticorpi este în continuare deficitară, se recomandă o doză de 0,5 g/kg şi lună până când nivelul de anticorpi revine la normal. Dozele recomandate sunt prezentate pe scurt în tabelul următor:
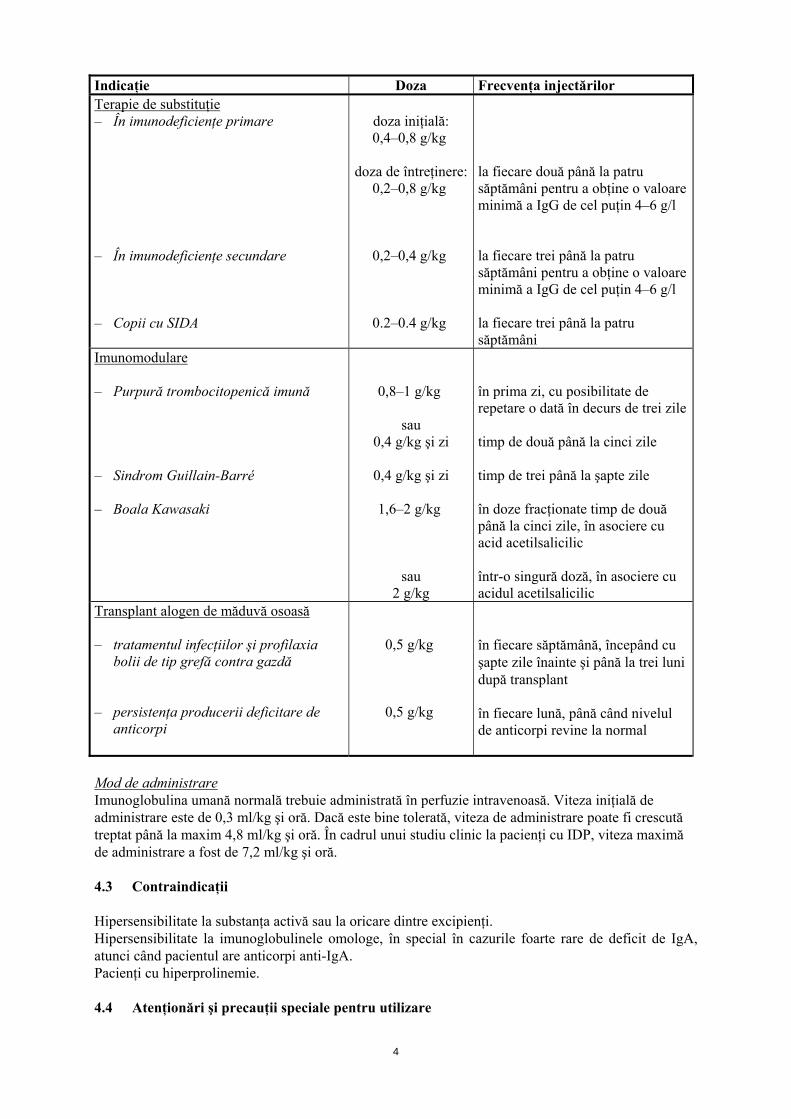
4
Indicaţie Doza Frecvenţa injectărilor Terapie de substituţie – În imunodeficienţe primare – În imunodeficienţe secundare – Copii cu SIDA
doza iniţială: 0,4–0,8 g/kg
doza de întreţinere:
0,2–0,8 g/kg
0,2–0,4 g/kg
0.2–0.4 g/kg
la fiecare două până la patru săptămâni pentru a obţine o valoare minimă a IgG de cel puţin 4–6 g/l la fiecare trei până la patru săptămâni pentru a obţine o valoare minimă a IgG de cel puţin 4–6 g/l la fiecare trei până la patru săptămâni
Imunomodulare – Purpură trombocitopenică imună – Sindrom Guillain-Barré – Boala Kawasaki
0,8–1 g/kg
sau 0,4 g/kg şi zi
0,4 g/kg şi zi
1,6–2 g/kg
sau 2 g/kg
în prima zi, cu posibilitate de repetare o dată în decurs de trei zile timp de două până la cinci zile timp de trei până la şapte zile în doze fracţionate timp de două până la cinci zile, în asociere cu acid acetilsalicilic într-o singură doză, în asociere cu acidul acetilsalicilic
Transplant alogen de măduvă osoasă – tratamentul infecţiilor şi profilaxia
bolii de tip grefă contra gazdă – persistenţa producerii deficitare de
anticorpi
0,5 g/kg
0,5 g/kg
în fiecare săptămână, începând cu şapte zile înainte şi până la trei luni după transplant în fiecare lună, până când nivelul de anticorpi revine la normal
Mod de administrare Imunoglobulina umană normală trebuie administrată în perfuzie intravenoasă. Viteza iniţială de administrare este de 0,3 ml/kg şi oră. Dacă este bine tolerată, viteza de administrare poate fi crescută treptat până la maxim 4,8 ml/kg şi oră. În cadrul unui studiu clinic la pacienţi cu IDP, viteza maximă de administrare a fost de 7,2 ml/kg şi oră. 4.3 Contraindicaţii Hipersensibilitate la substanţa activă sau la oricare dintre excipienţi. Hipersensibilitate la imunoglobulinele omologe, în special în cazurile foarte rare de deficit de IgA, atunci când pacientul are anticorpi anti-IgA. Pacienţi cu hiperprolinemie. 4.4 Atenţionări şi precauţii speciale pentru utilizare

5
Anumite reacţii adverse severe pot fi legate de viteza de administrare. Viteza de administrare recomandată la pct. 4.2 “Mod de administrare” trebuie respectată cu stricteţe. Pacienţii trebuie monitorizaţi atent, iar în timpul administrării trebuie observate toate simptomele. Anumite reacţii adverse pot apărea mai frecvent – în cazul unei viteze crescute de perfuzie, – la pacienţi cu hipo- sau agamaglobulinemie, cu sau fără deficit de IgA – la pacienţii cărora li se administrează pentru prima dată imunoglobulină umană normală sau, în
cazuri rare, când medicamentul conţinând imunoglobulină umană normală este schimbat cu altul sau când a trecut un interval lung de la ultima perfuzie.
Reacţiile reale de hipersensibilitate sunt rare. Acestea pot apărea în cazurile foarte rare de deficit de IgA cu anticorpi anti-IgA. Rareori, imunoglobulina umană normală poate induce o scădere a tensiunii arteriale cu reacţie anafilactică, chiar şi la pacienţi care au tolerat anterior tratamentul cu imunoglobulină umană normală. Potenţialele complicaţii pot fi adesea prevenite dacă se asigură că pacienţii: – nu prezintă sensibilitate la imunoglobulina umană normală, perfuzând la început medicamentul
foarte lent (0,3 ml/kg şi oră); – sunt monitorizaţi cu atenţie, observând orice simptom care apare în timpul perfuzării. În mod
special, pacienţii care nu au mai fost trataţi cu imunoglobulină umană normală, pacienţii cărora li s-a schimbat medicamentul conţinând Ig IV sau atunci când a trecut un interval lung de timp de la ultima administrare trebuie monitorizaţi în timpul perfuzării iniţiale şi în prima oră după prima perfuzie, pentru a observa eventualele reacţii adverse. Toţi ceilalţi pacienţi trebuie monitorizaţi timp de cel puţin douăzeci de minute după administrare.
Există dovezi clinice privind asocierea dintre administrarea de Ig IV şi evenimente tromboembolice cum sunt infarctul miocardic, accidentul vascular cerebral, embolia pulmonară şi tromboza venoasă profundă, care se presupune că este legată de o creştere relativă a vâscozităţii sângelui prin afluxul mare de imunoglobuline la pacienţii care prezintă risc. Prescrierea şi administrarea de Ig IV trebuie făcută cu prudenţă la pacienţii obezi şi la pacienţii cu factori de risc preexistenţi pentru apariţia unor evenimente tromboembolice (cum sunt vârsta înaintată, hipertensiunea arterială, diabetul zaharat şi antecedentele de afecţiuni vasculare sau episoade trombotice, pacienţii cu boli trombofilice dobândite sau ereditare, cei cu perioade prelungite de imobilizare, pacienţii cu hipovolemie severă şi pacienţii cu afecţiuni care cresc vâscozitatea sanguină). S-au raportat cazuri de insuficienţă renală acută la pacienţii trataţi cu Ig IV. În majoritatea cazurilor, s-au identificat factori de risc cum sunt: insuficienţă renală preexistentă, diabet zaharat, hipovolemie, obezitate, administrarea concomitentă a unor medicamente nefrotoxice sau vârsta peste 65 ani. În caz de insuficienţă renală, trebuie luată în considerare întreruperea tratamentului cu Ig IV. În timp ce aceste raportări de disfuncţie renală sau de insuficienţă renală acută s-au asociat cu administrarea mai multor medicamente pe bază de Ig IV autorizate, cele care conţin zahăr ca stabilizator s-au asociat cu o proporţie anormal de mare din numărul lor total. La pacienţii care prezintă risc poate fi luată în considerare utilizarea medicamentelor cu Ig IV care nu conţin zahăr. Privigen nu conţine zahăr sau alte tipuri de glucide. La pacienţii care prezintă risc de insuficienţă renală acută sau de reacţii adverse de tip tromboembolic, medicamentele cu Ig IV trebuie administrate în cea mai mică doză eficace şi perfuzate cu viteza minimă posibilă. În cazul tuturor pacienţilor, administrarea de Ig IV necesită: – o hidratare adecvată înaintea iniţierii perfuziei cu Ig IV – monitorizarea diurezei – monitorizarea creatininemiei – evitarea utilizării concomitente a diureticelor de ansă

6
În caz de reacţii adverse, trebuie redusă viteza de administrare sau trebuie oprită administrarea perfuziei. Tratamentul necesar depinde de natura şi severitatea reacţiei adverse. În caz de şoc trebuie instituit tratamentul medical standard pentru terapia şocului. Informaţii cu privire la riscul transmiterii agenţii infecţioşi Măsurile standard de prevenire a infecţiilor dobândite ca urmare a utilizării medicamentelor preparate din sânge sau plasmă umană includ selectarea donatorilor, testarea fiecărei probe donate şi a rezervei de plasmă pentru a evidenţia markerii specifici ai infecţiei şi introducerea în procesul de fabricaţie a unor etape eficiente pentru inactivarea/eliminarea virusurilor. Cu toate acestea, atunci când se administrează medicamente preparate din plasmă sau sânge uman, nu poate fi exclusă în totalitate posibilitatea de transmitere a unor agenţi infecţioşi. Aceasta se aplică şi în cazul virusurilor necunoscute sau noi şi a altor agenţi patogeni. Măsurile adoptate sunt considerate eficiente în cazul virusurilor încapsulate cum sunt HIV, VHB şi VHC şi pentru virusul neîncapsulat VHA şi virusul B19. Experienţa clinică nu a evidenţiat transmiterea hepatitei A sau a virusului B19 prin imunoglobuline şi se presupune, de asemenea, că o contribuţie importantă la siguranţa împotriva virusurilor o are conţinutul de anticorpi. Se recomandă ferm, ca de fiecare dată când se administrează Privigen unui pacient, să se înregistreze numele şi seria de fabricaţie pentru a menţine o legătură între pacient şi seria medicamentului. 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune Vaccinuri cu virusuri vii atenuate Administrarea de imunoglobuline poate diminua eficacitatea vaccinurilor cu virus viu atenuat cum sunt vaccinurile rujeolic, parotiditic, rubeolic şi varicelic pentru o perioadă de cel puţin şase săptămâni şi cel mult trei luni. După administrarea acestui medicament, trebuie să treacă un interval de trei luni înainte de vaccinarea cu vaccinuri cu virusuri vii atenuate. În cazul rujeolei, această perioadă de diminuare a eficacităţii vaccinului poate persista până la un an. De aceea, la pacienţii cărora li se administrează vaccin rujeolic trebuie să se verifice titrul anticorpilor. Interferenţa cu testele serologice După perfuzarea de imunoglobulină, creşterea tranzitorie în sângele pacienţilor a diverşilor anticorpi transferaţi pasiv poate determina obţinerea unor rezultate fals pozitive la testele serologice. Transmiterea pasivă a anticorpilor faţă de antigenele eritrocitare, de exemplu A, B, D, poate interfera cu anumite teste serologice pentru detectarea alo-anticorpilor eritrocitari (de exemplu testul Coombs).

7
4.6 Sarcina şi alăptarea Siguranţa utilizării acestui medicament în timpul sarcinii nu a fost stabilită prin studii clinice controlate şi, de aceea, trebuie administrat cu precauţie la femeile gravide şi la cele care alăptează. Experienţa clinică cu imunoglobuline sugerează că nu sunt de aşteptat efecte nocive asupra evoluţiei sarcinii sau asupra fătului şi nou-născutului. Imunoglobulinele se excretă în laptele matern şi pot contribui la transferul anticorpilor protectori la nou-născut. 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje Nu s-au observat efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje. 4.8 Reacţii adverse Ocazional, după administrarea intravenoasă de imunoglobulină umană normală, pot apărea reacţii adverse cum sunt: frisoane, cefalee, febră, vărsături, reacţii alergice, greaţă, artralgii, hipotensiune arterială şi lombalgii moderate. Rareori, imunoglobulinele umane normale pot determina o scădere bruscă a tensiunii arteriale şi, în cazuri izolate, şoc anafilactic, chiar dacă pacientul nu a prezentat hipersensibilitate la administrările anterioare. După administrarea de imunoglobulină umană normală s-au observat cazuri de meningită aseptică reversibilă, cazuri izolate de hemoliză/anemie hemolitică reversibilă şi cazuri rare de reacţii cutanate tranzitorii. S-au observat creşteri ale creatininemiei şi/sau insuficienţă renală acută. Foarte rar: Reacţii tromboembolice cum sunt infarct miocardic, accident vascular cerebral, embolie pulmonară şi tromboze venoase profunde. S-au efectuat două studii clinice cu Privigen, unul la pacienţi cu imunodeficienţă primară (IDP) şi unul la pacienţi cu purpură trombocitopenică imună (PTI). În studiul asupra IDP, 80 de subiecţi au fost înrolaţi şi trataţi cu Privigen. Dintre aceştia, 72 au încheiat cele douăsprezece luni de tratament. Studiul asupra PTI a fost efectuat la 57 de pacienţi. Majoritatea reacţiilor adverse la medicament (RAM) observate în cele două studii clinice au fost uşoare până la moderate. RAM raportate în cele două studii sunt prezentate în tabelul de mai jos conform clasificării MedDRA pe aparate, sisteme şi organe, şi în funcţie de frecvenţa de apariţie. Frecvenţa a fost evaluată utilizând următoarele criterii: foarte frecvente (≥1/10), frecvente (≥1/100 şi <1/10), mai puţin frecvente (≥1/1000 şi <1/100). În cadrul fiecărei grupe de frecvenţă, reacţiile adverse sunt prezentate în ordinea descrescătoare a gravităţii.
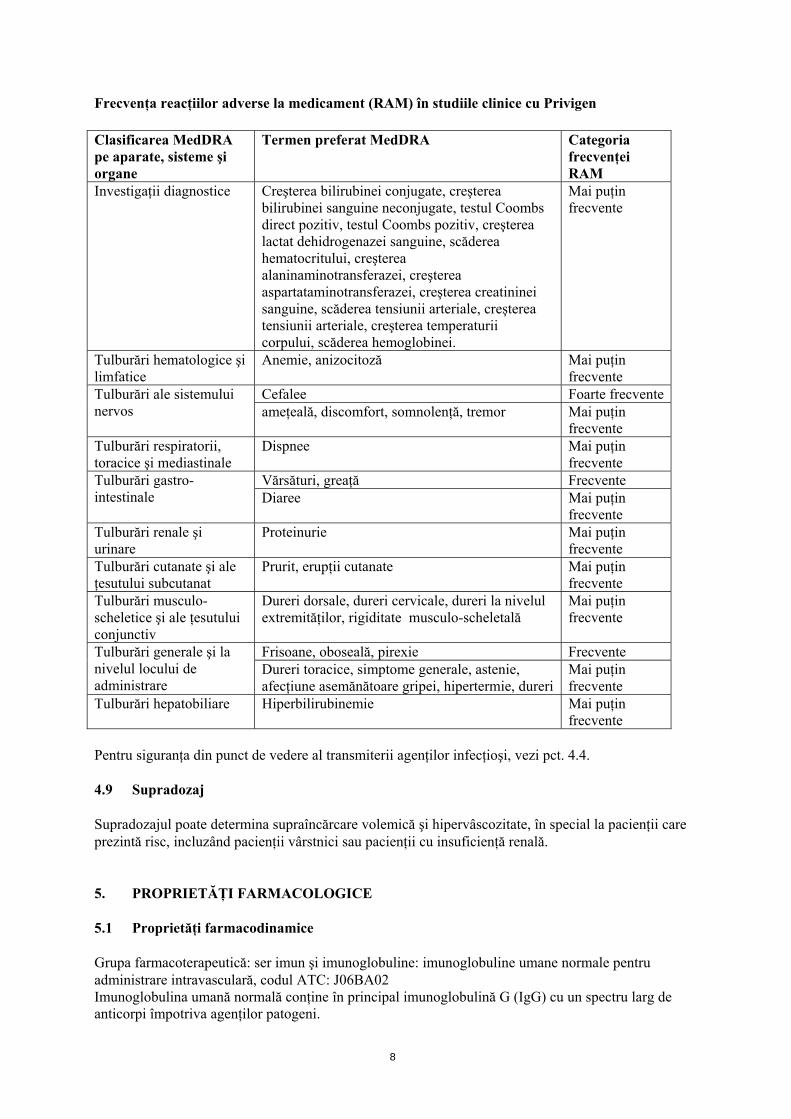
8
Frecvenţa reacţiilor adverse la medicament (RAM) în studiile clinice cu Privigen Clasificarea MedDRA pe aparate, sisteme şi organe
Termen preferat MedDRA Categoria frecvenţei RAM
Investigaţii diagnostice Creşterea bilirubinei conjugate, creşterea bilirubinei sanguine neconjugate, testul Coombs direct pozitiv, testul Coombs pozitiv, creşterea lactat dehidrogenazei sanguine, scăderea hematocritului, creşterea alaninaminotransferazei, creşterea aspartataminotransferazei, creşterea creatininei sanguine, scăderea tensiunii arteriale, creşterea tensiunii arteriale, creşterea temperaturii corpului, scăderea hemoglobinei.
Mai puţin frecvente
Tulburări hematologice şi limfatice
Anemie, anizocitoză Mai puţin frecvente
Cefalee Foarte frecventeTulburări ale sistemului nervos ameţeală, discomfort, somnolenţă, tremor Mai puţin
frecvente Tulburări respiratorii, toracice şi mediastinale
Dispnee Mai puţin frecvente
Vărsături, greaţă Frecvente Tulburări gastro-intestinale Diaree Mai puţin
frecvente Tulburări renale şi urinare
Proteinurie Mai puţin frecvente
Tulburări cutanate şi ale ţesutului subcutanat
Prurit, erupţii cutanate Mai puţin frecvente
Tulburări musculo-scheletice şi ale ţesutului conjunctiv
Dureri dorsale, dureri cervicale, dureri la nivelul extremităţilor, rigiditate musculo-scheletală
Mai puţin frecvente
Frisoane, oboseală, pirexie Frecvente Tulburări generale şi la nivelul locului de administrare
Dureri toracice, simptome generale, astenie, afecţiune asemănătoare gripei, hipertermie, dureri
Mai puţin frecvente
Tulburări hepatobiliare Hiperbilirubinemie Mai puţin frecvente
Pentru siguranţa din punct de vedere al transmiterii agenţilor infecţioşi, vezi pct. 4.4. 4.9 Supradozaj Supradozajul poate determina supraîncărcare volemică şi hipervâscozitate, în special la pacienţii care prezintă risc, incluzând pacienţii vârstnici sau pacienţii cu insuficienţă renală. 5. PROPRIETĂŢI FARMACOLOGICE 5.1 Proprietăţi farmacodinamice Grupa farmacoterapeutică: ser imun şi imunoglobuline: imunoglobuline umane normale pentru administrare intravasculară, codul ATC: J06BA02 Imunoglobulina umană normală conţine în principal imunoglobulină G (IgG) cu un spectru larg de anticorpi împotriva agenţilor patogeni.

9
Imunoglobulina umană normală conţine anticorpi IgG prezenţi la populaţia normală. De obicei, este preparată din rezerve de plasmă de la minim 1000 donatori. Conţine o distribuţie a subclaselor de imunoglobulină G proporţională cu cea din plasma umană nativă. Dozele adecvate din acest medicament pot restabili la valori normale concentraţiile de imunoglobulină G scăzute în mod patologic. Mecanismul de acţiune pentru alte indicaţii decât terapia de substituţie nu este complet elucidat, dar include efecte imunomodulatoare. Siguranţa şi eficacitatea medicamentului Privigen au fost evaluate în două studii prospective, deschise, cu un singur braţ, multicentrice efectuate în Europa (studiul PTI), respectiv în Europa şi SUA (studiul IDP). 5.2 Proprietăţi farmacocinetice După administrarea intravenoasă, imunoglobulina umană normală este biodisponibilă imediat şi în totalitate în circulaţia primitorului. Se distribuie relativ rapid în plasmă şi în lichidul extravascular; după aproximativ trei până la cinci zile se realizează un echilibru între compartimentele intra- şi extravascular. Parametrii farmacocinetici pentru Privigen s-au determinat într-un studiu clinic la pacienţi cu IDP (vezi pct. 5.1). Douăzecişicinci de pacienţi (cu vârste cuprinse între 13 şi 69 de ani) au participat la evaluările farmacocinetice (vezi tabelul de mai jos). Timpul mediu de înjumătăţire plasmatică al medicamentului Privigen la pacienţii cu imunodeficienţă primară a fost de 36,6 zile. Acest timp de înjumătăţire poate varia individual, în special în imunodeficienţa primară. Parametrii farmacocinetici ai medicamentului Privigen la 25 de pacienţi cu IDP Parametru Mediană (limite) Cmax (valoare maximă, g/l) 23,4 (10,4-34,6) Cmin (valoare minimă, g/l) 10,2 (5,8-14,7) t½ (zile) 36,6 (20,6-96,6) Cmax, concentraţia serică maximă; Cmin, concentraţia serică minimă (valoarea minimă); t½, timpul de înjumătăţire prin eliminare IgG şi complexele IgG sunt distruse în celule de către sistemul reticuloendotelial. 5.3 Date preclinice de siguranţă Imunoglobulinele sunt componente normale ale organismului uman. L-prolina este un aminoacid neesenţial, fiziologic. Siguranţa medicamentului Privigen a fost evaluată în câteva studii preclince, referitoare în special la excipientul L-prolină. Câteva studii publicate referitoare la hiperprolinemie au evidenţiat faptul că dozele mari de L-prolină administrate pe perioade îndelungate au efecte asupra dezvoltării creierului la şobolanii foarte tineri. Cu toate acestea, în studiile în care doza administrată a reflectat indicaţiile clinice pentru Privigen, nu s-a observat niciun efect asupra dezvoltării creierului. Datele non-clinice nu evidenţiază riscuri speciale pentru om pe baza studiilor farmacologice şi toxicologice de siguranţă. 6. PROPRIETĂŢI FARMACEUTICE 6.1 Lista excipienţilor L-prolină

10
Apă pentru preparate injectabile 6.2 Incompatibilităţi Acest medicament nu trebuie amestecat cu alte medicamente, cu excepţia celor menţionate la punctul 6.6. 6.3 Perioada de valabilitate 2 ani 6.4 Precauţii speciale pentru păstrare A nu se păstra la temperaturi peste 25°C. A nu se congela. A se păstra flaconul în cutie pentru a fi protejat de lumină. 6.5 Natura şi conţinutul ambalajului 50 sau 100 ml soluţie într-un flacon (sticlă de tip I sau II) cu dop (elastomer), capac (capsă din aluminiu), disc detaşabil (plastic), etichetă cu agăţător integrat. 200 ml soluţie într-un flacon (sticlă de tip II), cu dop (elastomer), capac (capsă din aluminiu), disc detaşabil (plastic), etichetă cu agăţător integrat. Mărimea ambalajului: 1 flacon Este posibil ca nu toate mărimile de ambalaj să fie comercializate. 6.6 Precauţii speciale pentru eliminarea reziduurilor Privigen se livrează ca soluţie gata preparată în flacoane de unică folosinţă. Înainte de utilizare, medicamentul trebuie adus la temperatura camerei sau la temperatura corpului. Pentru administrarea medicamentului Privigen se va folosi o linie de perfuzie cu supapă. Înţepaţi întotdeauna dopul în centru, în interiorul marcajului. Soluţia trebuie să fie limpede sau uşor opalescentă. A nu se utiliza soluţiile tulburi sau care prezintă particule. Odată ce flaconul a fost înţepat în condiţii aseptice, conţinutul se va folosi imediat. Deoarece soluţia nu conţine conservanţi, Privigen trebuie perfuzat cât mai curând posibil. Orice produs neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale. 7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ CSL Behring GmbH Emil-von-Behring-Strasse 76 D-35041 Marburg Germania 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ

11
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI 10. DATA REVIZUIRII TEXTULUI Informaţii detaliate despre acest produs sunt disponibile pe website-ul Agenţiei Europene a Medicamentului (EMEA): http;//www.emea.europa.eu/

12
ANEXA II
A. PRODUCĂTORUL SUBSTANŢEI BIOLOGIC ACTIVE ŞI DEŢINĂTORII AUTORIZAŢIEI DE FABRICAŢIE RESPONSABILI PENTRU ELIBERAREA SERIEI
B. CONDIŢIILE EMITERII AUTORIZAŢIEI DE PUNERE PE PIAŢĂ

13
A. PRODUCĂTORUL SUBSTANŢEI BIOLOGIC ACTIVE ŞI DEŢINĂTORII AUTORIZAŢIEI DE FABRICAŢIE RESPONSABILI PENTRU ELIBERAREA SERIEI
Numele şi adresa producătorului(ilor) substanţei(lor) biologic active CSL Behring AG Wankdorfstrasse 10, 3000 Bern 22 Elveţia Numele şi adresa producătorului(ilor) responsabil(i) pentru eliberarea seriei CSL Behring GmbH Emil-von-Behring-Strasse 76 D-35041 Marburg Germania Prospectul tipărit al medicamentului trebuie să menţioneze numele şi adresa producǎtorului responsabil pentru eliberarea seriei respective. B. CONDIŢIILE EMITERII AUTORIZAŢIEI DE PUNERE PE PIAŢĂ • CONDIŢII SAU RESTRICŢII PRIVIND FURNIZAREA ŞI UTILIZAREA IMPUSE
DEŢINĂTORULUI AUTORIZAŢIEI DE PUNERE PE PIAŢĂ Medicament cu eliberare pe bază de prescripţie medicală. • ALTE CONDIŢII Sistem de farmacovigilenţă DAPP trebuie să se asigure că sistemul de farmacovigilenţă, aşa cum este descris în versiunea 1.0 prezentată în modulul 1.8.1. al cererii de autorizaţie de punere pe piaţă, există şi este funcţional înainte şi în timpul prezenţei medicamentului pe piaţă. Plan de Management al Riscului DAPP se angajează să efectueze studiile şi activităţile suplimentare legate de farmacovigilenţă descrise în detaliu în planul de farmacovigilenţă, aşa cum s-a stabilit în versiunea 1.3 a Planului de Management al Riscului (PMR) prezentat în modulul 1.8.2. al cererii de autorizaţie de punere pe piaţă, precum şi în orice actualizări ulterioare ale PMR aprobate de Comitetul pentru medicamente de uz uman (CHMP). Conform recomandărilor CHMP cu privire la sistemele de management al riscului pentru medicamente de uz uman, PMR-ul actualizat trebuie depus odată cu următorul Raport Periodic Actualizat privind Siguranţa (RPAS). În plus, trebuie înaintat un PMR actualizat • Dacă se primesc informaţii noi care pot avea un impact asupra specificaţiei de siguranţă actuale,
planului de farmacovigilenţă sau activităţilor de reducere la minim a riscului • În decurs de 60 de zile de la realizarea unui obiectiv important (farmacovigilenţă sau reducerea
la minim a riscului) • La cererea EMEA Eliberarea oficială a seriei: în conformitate cu articolul 114 al Directivei 2001/83/EG revizuite, eliberarea oficială a seriei se va face de către un laborator de stat sau un laborator desemnat în acest scop.

14
ANEXA III
ETICHETAREA ŞI PROSPECTUL

15
A. ETICHETAREA

16
INFORMAŢII CARE TREBUIE SĂ APARĂ PE AMBALAJUL SECUNDAR CUTIE 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI Privigen 100 mg/ml soluţie perfuzabilă Imunoglobulină umană normală (Ig IV) 2. DECLARAREA SUBSTANŢEI(LOR) ACTIVE Un ml conţine: Proteină plasmatică umană.......100 mg Puritate IgG .............................. ≥ 98% IgA.....................................≤ 0,025 mg 5 g/50 ml 10 g/100 ml 20 g/200 ml Se vor nota în colţul din dreapta sus al feţei principale a cutiei pentru a reda conţinutul total şi volumul flaconului 3. LISTA EXCIPIENŢILOR Excipienţi: L-prolină, apă pentru preparate injectabile. 4. FORMA FARMACEUTICĂ ŞI CONŢINUTUL Soluţie perfuzabilă (10%) Conţine 1 flacon. 5. MODUL ŞI CALEA(CĂILE) DE ADMINISTRARE Administrare intravenoasă. A se citi prospectul înainte de utilizare. 6. ATENŢIONARE SPECIALĂ PRIVIND FAPTUL CĂ MEDICAMENTUL NU TREBUIE
PĂSTRAT LA ÎNDEMÂNA ŞI VEDEREA COPIILOR A nu se lăsa la îndemâna şi vederea copiilor. 7. ALTĂ(E) ATENŢIONARE(ĂRI) SPECIALĂ(E), DACĂ ESTE(SUNT) NECESARĂ(E) 8. DATA DE EXPIRARE EXP

17
9. CONDIŢII SPECIALE DE PĂSTRARE A nu se păstra la temperaturi peste 25°C. A nu se congela. A se păstra flaconul în cutie pentru a fi protejat de lumină. 10. PRECAUŢII SPECIALE PRIVIND ELIMINAREA MEDICAMENTELOR
NEUTILIZATE SAU A MATERIALELOR REZIDUALE PROVENITE DIN ASTFEL DE MEDICAMENTE, DACĂ ESTE CAZUL
11. NUMELE ŞI ADRESA DEŢINĂTORULUI AUTORIZAŢIEI DE PUNERE PE PIAŢĂ Deţinătorul autorizaţiei de punere pe piaţă: CSL Behring GmbH Emil-von-Behring-Strasse 76 D-35041 Marburg Germania 12. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ 13. SERIA DE FABRICAŢIE LOT 14. CLASIFICARE GENERALĂ PRIVIND MODUL DE ELIBERARE Medicament eliberat pe bază de prescripţie medicală. 15. INSTRUCŢIUNI DE UTILIZARE 16. INFORMAŢII ÎN BRAILLE Justificare acceptată pentru neincluderea informaţiei în Braille.

18
INFORMAŢII CARE TREBUIE SĂ APARĂ PE AMBALAJUL PRIMAR FLACON 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI Privigen 100 mg/ml soluţie perfuzabilă Imunoglobulină umană normală (Ig IV) 2. DECLARAREA SUBSTANŢEI(LOR) ACTIVE Un ml conţine: Proteină plasmatică umană: 100 mg; puritate IgG: ≥ 98%. IgA: ≤ 0,025 mg. 5 g/50 ml 10 g/100 ml 20 g/200 ml Vor fi notate în colţul din dreapta sus al etichetei pentru a reda conţinutul total şi volumul flaconului 3. LISTA EXCIPIENŢILOR L-prolină, apă pentru preparate injectabile. 4. FORMA FARMACEUTICĂ ŞI CONŢINUTUL Soluţie perfuzabilă (10%) 5. MODUL ŞI CALEA(CĂILE) DE ADMINISTRARE Administrare intravenoasă. A se citi prospectul înainte de utilizare. 6. ATENŢIONARE SPECIALĂ PRIVIND FAPTUL CĂ MEDICAMENTUL NU TREBUIE
PĂSTRAT LA ÎNDEMÂNA ŞI VEDEREA COPIILOR A nu se lăsa la îndemâna şi vederea copiilor. 7. ALTĂ(E) ATENŢIONARE(ĂRI) SPECIALĂ(E), DACĂ ESTE(SUNT) NECESARĂ(E) 8. DATA DE EXPIRARE EXP 9. CONDIŢII SPECIALE DE PĂSTRARE A nu se păstra la temperaturi peste 25 °C. A nu se congela.

19
A se păstra flaconul în cutie pentru a fi protejat de lumină. 10. PRECAUŢII SPECIALE PRIVIND ELIMINAREA MEDICAMENTELOR
NEUTILIZATE SAU A MATERIALELOR REZIDUALE PROVENITE DIN ASTFEL DE MEDICAMENTE, DACĂ ESTE CAZUL
11. NUMELE ŞI ADRESA DEŢINĂTORULUI AUTORIZAŢIEI DE PUNERE PE PIAŢĂ CSL Behring GmbH, D-35041 Marburg, Germania 12. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ 13. SERIA DE FABRICAŢIE LOT 14. CLASIFICARE GENERALĂ PRIVIND MODUL DE ELIBERARE Medicament eliberat pe bază de prescripţie medicală. 15. INSTRUCŢIUNI DE UTILIZARE 16. INFORMAŢII ÎN BRAILLE Justificare acceptată pentru neincluderea informaţiei în Braille.

20
B. PROSPECTUL

21
PROSPECT: INFORMAŢII PENTRU UTILIZATOR
Privigen 100 mg/ml (10%) soluţie perfuzabilă Imunoglobulină umană normală (Ig IV)
Citiţi cu atenţie şi în întregime acest prospect înainte de a începe să utilizaţi acest medicament - Păstraţi acest prospect. S-ar putea să fie necesar să-l recitiţi. - Dacă aveţi orice întrebări suplimentare, adresaţi-vă medicului dumneavoastră sau farmacistului. - Dacă vreuna dintre reacţiile adverse devine gravă sau dacă observaţi orice reacţie adversă
nemenţionată în acest prospect, vă rugăm să-i spuneţi medicului dumneavoastră sau farmacistului.
În acest prospect găsiţi: 1. Ce este Privigen şi pentru ce se utilizează 2. Înainte să utilizaţi Privigen 3. Cum să utilizaţi Privigen 4. Reacţii adverse posibile 5. Cum se păstrează Privigen 6. Informaţii suplimentare 1. CE ESTE PRIVIGEN ŞI PENTRU CE SE UTILIZEAZĂ Ce este Privigen Privigen este o soluţie perfuzabilă gata preparată. Soluţia conţine proteine speciale, izolate din plasma sanguină umană. Aceste proteine fac parte din clasa “imunoglobulinelor”, denumite de asemenea anticorpi. Cum acţionează Privigen Anticorpii sunt de obicei produşi de sistemul nostru imunitar şi ajută organismul să lupte împotriva infecţiilor. Anumite boli pot provoca tulburări severe ale sistemului imunitar. Din această cauză s-ar putea să nu aveţi suficienţi anticorpi proprii sau să aveţi nevoie de anticorpi suplimentari. Anticorpii care vă sunt administraţi prin Privigen pot completa numărul de anticorpi proprii sau pot substitui anticorpii care lipsesc. Anticorpii din Privigen sunt izolaţi din plasmă sanguină umană. Din acest motiv acţionează ca şi cum ar fi proprii dumneavoastră anticorpi. De asemenea, Privigen poate reduce simptomele anumitor afecţiuni inflamatorii. În aceste cazuri Privigen reglează sistemul imunitar care este tulburat. Efectele acestea nu sunt însă înţelese în întregime. Pentru ce se utilizează Privigen Privigen se utilizează în trei situaţii diferite: A) Tratamentul pacienţilor care nu au o cantitate suficientă de anticorpi (terapie de substituţie).
Aceştia se împart în trei grupe: 1. Pacienţi cu deficit înnăscut de anticorpi (sindroame de imunodeficienţă primară (IDP))
cum sunt: • agamaglobulinemie sau hipogamaglobulinemie congenitale, • imunodeficienţe comune variabile, • imunodeficienţe combinate severe, • sindromul Wiskott Aldrich.

22
2. Pacienţi cu anumite tipuri de cancer de sânge care determină o producţie deficitară de anticorpi şi o tendinţă la infecţii recurente, precum: • mielomul, • leucemia limfocitară cronică cu hipogamaglobulinemie secundară severă.
3. Copii cu SIDA (Sindrom Imunodeficitar Dobândit) congenitală şi cu infecţii recurente. B) Tratamentul pacienţilor cu anumite afecţiuni inflamatorii (imunomodulare). Aceştia se împart în
trei grupe: 1. Pacienţi cu număr insuficient de trombocite (purpură trombocitopenică imună (PTI)), şi
• care au risc crescut de sângerare, • care urmează să fie supuşi unei intervenţii chirurgicale în viitorul apropiat.
2. Pacienţi cu sindromul Guillain Barré. Acesta este o afecţiune acută caracterizată prin inflamaţia nervilor periferici care determină astenie musculară severă, în principal la nivelul membrelor inferioare şi superioare.
3. Pacienţi cu boala Kawasaki. Aceasta este o afecţiune acută întâlnită îndeosebi la copiii mici, caracterizată printr-o inflamaţie a vaselor de sânge din întreg organismul.
C) Tratamentul sau prevenirea infecţiilor după un transplant de măduvă osoasă (transplant alogen
de măduvă osoasă). 2. ÎNAINTE SĂ UTILIZAŢI PRIVIGEN
Vă rugăm să citiţi cu atenţie acest punct. Informaţiile pe care le conţine trebuie luate în considerare de dumneavoastră şi de medicul dumneavoastră, înainte să utilizaţi Privigen.
Privigen nu trebuie utilizat Dacă sunteţi alergic (hipersensibil)
la imunoglobuline umane la oricare dintre celelalte componente ale Privigen (pentru lista completă a excipienţilor
vezi punctul 6 al acestui prospect). Vă rugăm să informaţi medicul dumneavoastră sau personalul medical înainte de
începerea tratamentului, cu privire la orice medicament sau aliment pe care nu l-aţi tolerat.
Dacă în sângele dumneavoastră sunt prezenţi anticorpi împotriva imunoglobulinelor de tip IgA. Această situaţie se întâlneşte foarte rar şi poate surveni dacă nu aveţi o cantitate suficientă de
imunoglobuline de tip IgA în sânge. Vă rugăm să informaţi medicul dumneavoastră sau personalul medical înainte de
începerea tratamentului, dacă aveţi un deficit de imunoglobuline de tip IgA. Dacă în sângele dumneavoastră se găseşte o cantitate prea mare din aminoacidul prolină
(hiperprolinemie). Această afecţiune este extrem de rară. În toată lumea sunt cunoscute doar câteva familii cu această afecţiune.
Vă rugăm să informaţi medicul dumneavoastră sau personalul medical înainte de începerea tratamentului, dacă aveţi o cantitate prea mare de prolină în sânge.
Aveţi grijă deosebită când utilizaţi Privigen Riscul apariţiei anumitor reacţii adverse poate creşte în următoarele situaţii:
sunteţi supraponderal, sunteţi în vârstă, aveţi diabet zaharat, aţi fost imobilizat la pat pentru o perioadă mai îndelungată, aveţi sau aţi avut deja probleme cu vasele de sânge (boli vasculare sau obstruarea unui vas
de sânge), aveţi sau aţi avut deja probleme cu rinichii,

23
aveţi tensiunea arterială crescută, volumul sanguin este prea mic (hipovolemie), suferiţi de o boală care determină îngroşarea sângelui, manifestaţi o tendinţă crescută de coagulare a sângelui (trombofilie), suferiţi de o boală care determină valori scăzute ale anticorpilor în sânge,
(hipogamaglobulinemie sau agamaglobulinemie), suferiţi de o boală renală, folosiţi medicamente care pot afecta rinichii (medicamente nefrotice), utilizaţi Privigen pentru prima dată sau după o pauză de tratament îndelungată (de ex. mai
multe luni). Vă rugăm să informaţi medicul dumneavoastră sau personalul medical înainte de
începerea tratamentului, dacă cel puţin una dintre aceste situaţii se aplică în cazul dumneavoastră. Medicul dumneavoastră va alege în acest caz imunoglobulina potrivită pentru administrare intravenoasă şi va lua măsuri speciale de precauţie.
Puteţi fi alergic (hipersensibil) la imunoglobuline (anticorpi) fără să ştiţi.
Acest fapt poate surveni chiar dacă aţi mai utilizat înainte imunoglobuline umane şi le-aţi tolerat. Se poate întâmpla îndeosebi dacă nu aveţi o cantitate suficientă de imunoglobuline de tip IgA în sânge. În aceste cazuri rare pot apărea reacţii alergice cum sunt scăderea bruscă a tensiunii arteriale sau şoc.
Dacă observaţi astfel de reacţii în timpul perfuziei cu Privigen, anunţaţi imediat medicul. Acesta va decide dacă este necesară încetinirea ritmului de perfuzare sau oprirea administrării perfuziei.
Pentru siguranţa dumneavoastră personală, tratamentul cu Privigen se va face sub supravegherea medicului dumneavoastră sau a personalului medical. De obicei veţi fi supravegheat în timpul perfuziei şi timp de cel puţin 20 de minute după oprirea acesteia. În anumite situaţii pot fi necesare măsuri speciale de precauţie. Câteva exemple de astfel de situaţii sunt: vi se administrează Privigen cu o viteză crescută de perfuzare sau vi se administrează Privigen prima dată sau a trecut un interval lung de timp (de exemplu câteva
luni) de când l-aţi utilizat ultima dată. În asemenea cazuri, veţi fi monitorizat cu atenţie în timpul perfuziei şi timp de cel puţin o oră după ce s-a oprit perfuzarea. Informaţii despre provenienţa substanţelor conţinute în Privigen Privigen este alcătuit din plasmă umană (partea lichidă a sângelui). În cazul medicamentelor preparate din sânge sau plasmă umană, se adoptă anumite măsuri pentru a preveni transmiterea infecţiilor la pacienţi. Acestea includ: selectarea atentă a donatorilor de sânge şi plasmă pentru excluderea celor cu risc de transmitere
a infecţiilor, şi testarea fiecărei probe donate şi a rezervei de plasmă pentru evidenţierea existenţei
virusurilor/infecţiilor. De asemenea, în cursul procesării sângelui şi plasmei, producătorii acestor medicamente includ etape care au rol de inactivare sau eliminare a virusurilor. În pofida acestor măsuri, atunci când se administrează medicamente preparate din plasmă sau sânge uman, nu poate fi exclusă în totalitate posibilitatea de transmitere a unor agenţi infecţioşi. Acest lucru este valabil şi în cazul virusurilor necunoscute sau nou apărute, precum şi al altor tipuri de infecţii. Măsurile adoptate sunt considerate eficace în cazul virusurilor încapsulate cum sunt virusul imunodeficienţei umane (HIV), virusurile hepatitice B şi C, şi al virusurilor neîncapsulate, cum sunt virusurile hepatitice A şi B19. Imunoglobulinele ca Privigen nu au fost asociate cu hepatite A sau B19. Aceasta se datorează probabil faptului că anticorpii care luptă împotriva acestor infecţii sunt prezenţi şi în imunoglobuline. Este posibil ca aceşti anticorpi să ajute la prevenirea hepatitelor A şi B19.

24
Se recomandă ferm, ca de fiecare dată când vi se administrează Privigen, să se înregistreze numele şi seria de fabricaţie a medicamentului pentru a se menţine o evidenţă a seriilor folosite.
Utilizarea Privigen cu alte medicamente
Vă rugăm să anunţaţi medicul dumneavoastră sau personalul medical înainte de începerea tratamentului dacă utilizaţi în prezent alte medicamente sau dacă aţi utilizat recent alte medicamente. Acestea includ şi medicamentele eliberate fără prescripţie medicală.
Vaccinuri Administrarea de Privigen poate diminua eficacitatea anumitor vaccinuri. Este vorba de vaccinurile cu virus viu atenuat cum sunt vaccinurile împotriva rujeolei, oreionului, rubeolei şi varicelei. Astfel de vaccinări trebuie amânate cu cel puţin 3 luni după administrarea ultimei perfuzii cu Privigen. În cazul rujeolei, această perioadă de diminuare a eficacităţii vaccinului poate persista până la 1 an. De aceea, medicul care administrează vaccinul trebuie să verifice eficacitatea vaccinului rujeolic.
Vă rugăm să informaţi înainte de vaccinare, medicul care administrează vaccinul, cu privire la tratamentul cu Privigen.
Teste sanguine După perfuzarea de Privigen, rezultatele anumitor teste sanguine (testele serologice) pot fi eronate pentru o anumită perioadă de timp.
Vă rugăm să informaţi medicul cu privire la tratamentul cu Privigen, înainte de efectuarea oricărui test sanguin.
Sarcina şi alăptarea
Vă rugăm să informaţi medicul dumneavoastră sau personalul medical dacă sunteţi gravidă sau alăptaţi. Medicul dumneavoastră va decide dacă puteţi utiliza Privigen în timpul sarcinii sau alăptării.
Utilizarea medicamentului Privigen la femeile gravide şi la cele care alăptează nu a fost studiată în mod separat. Cu toate acestea, s-au utilizat medicamente care conţin anticorpi la femei gravide sau care alăptează. Experienţa îndelungată a demonstrat că nu există efecte nocive asupra evoluţiei sarcinii sau asupra fătului. Dacă alăptaţi şi urmaţi tratament cu Privigen, anticorpii medicamentului vor fi regăsiţi în laptele matern. În acest fel, şi copilul poate primi anticorpi protectori. Conducerea vehiculelor şi folosirea utilajelor Privigen nu are nicio influenţă asupra capacităţii de a conduce vehicule sau de a folosi utilaje. 3. CUM SĂ UTILIZAŢI PRIVIGEN Privigen se administrează de obicei de către medicul dumneavoastră sau asistenta medicală. Privigen este destinat doar perfuzării într-o venă (perfuzie intravenoasă). Medicul dumneavoastră decide ce cantitate de Privigen veţi utiliza. Cantitatea depinde de
afecţiune, starea dumneavoastră clinică şi de greutatea dumneavoastră corporală. La începutul administrării, Privigen se perfuzează cu viteză mică. În funcţie de cum vă simţiţi,
medicul dumneavoastră poate creşte ulterior viteza administrării. Dacă utilizaţi o cantitate mai mare de Privigen decât este necesar

25
Privigen se administrează de regulă doar sub supraveghere medicală. Astfel, este foarte puţin probabil să apară supradozaj. În pofida acestui fapt, dacă utilizaţi o cantitate mai mare de Privigen decât este necesar, sângele dumneavoastră poate deveni prea gros (hipervâscos). Aceasta se poate întâmpla în special dacă sunteţi un pacient cu risc, de exemplu un pacient în vârstă sau cu boli renale. 4. REACŢII ADVERSE POSIBILE
Ca toate medicamentele, Privigen poate provoca reacţii adverse, cu toate că acestea nu apar la toate persoanele. Puteţi fi alergic (hipersensibil) la imunoglobuline (anticorpi) şi pot apărea reacţii alergice cum
sunt scăderea bruscă a tensiunii arteriale sau şoc. Dacă observaţi astfel de reacţii în timpul perfuziei cu Privigen, vă rugăm anunţaţi
imediat medicul dumneavoastră. Citiţi de asemenea punctul 2 al acestui prospect, referitor la riscul apariţiei reacţiilor alergice.
Apariţia reacţiilor adverse posibile poate fi redusă sau chiar evitată printr-o viteză de
administrare scăzută a medicamentului Privigen. Experienţa generală cu medicamente conţinând imunoglobulină a demonstrat că pot apărea următoarele reacţii adverse: dureri de cap, frisoane, febră, vărsături, reacţii uşoare de hipersensibilitate (reacţii alergice), greaţă, dureri articulare (artralgii), tensiune arterială scăzută, dureri moderate de spate În cazuri rare şi izolate, s-au raportat de asemenea următoarele reacţii adverse la medicamente conţinând imunoglobulină: meningită temporară aseptică (meningită aseptică reversibilă), reacţii cutanate tranzitorii creşterea valorii creatininei din sânge scăderea bruscă a tensiunii arteriale insuficienţă renală acută reacţii de hipersensibilitate severe (şoc anafilactic), chiar dacă nu aţi manifestat
hipersensibilitate la perfuziile anterioare formarea de cheaguri de sâge care pot ajunge în sistemul circulator sanguin (reacţii
tromboembolice) şi care pot determina, de exemplu: – infarct miocardic – accident vascular cerebral – obstruarea unui vas de sânge la nivel pulmonar (embolie pulmonară) – tromboză venoasă profundă
scădere temporară a globulelor roşii (anemie hemolitică reversibilă/hemoliză). Astfel de reacţii adverse pot apărea chiar dacă vi s-au administrat în trecut imunoglobuline umane (anticorpi) şi le-aţi tolerat.
Vă rugăm să spuneţi medicului dumneavoastră sau personalului medical dacă vreuna dintre reacţiile adverse devine gravă sau dacă observaţi orice reacţie adversă nemenţionată în acest prospect.

26
5. CUM SE PĂSTREAZĂ PRIVIGEN A nu se lăsa la îndemâna şi vederea copiilor. Nu utilizaţi Privigen după data de expirare înscrisă pe cutie şi eticheta flaconului, după EXP. Data de expirare se referă la ultima zi a lunii respective. A nu se păstra la temperaturi peste 25 °C. A nu se congela. A se păstra flaconul în cutie pentru a fi protejat de lumină. Nu utilizaţi Privigen dacă observaţi că soluţia este tulbure sau prezintă particule. 6. INFORMAŢII SUPLIMENTARE Ce conţine Privigen Substanţa activă este imunoglobulina umană normală (anticorpi de tipul IgG). Privigen conţine
100 mg/ml (10%) proteină umană din care minim 98% este IgG. Celelalte componente sunt aminoacidul L-prolină şi apa pentru preparate injectabile. Cum arată Privigen şi conţinutul ambalajului Privigen se prezintă sub formă de soluţie perfuzabilă. Soluţia este limpede sau uşor opalescentă, incoloră până la galben pal. Mărimea ambalajului este de 1 flacon (5 g/50 ml, 10 g/100 ml sau 20 g/200 ml). Este posibil ca nu toate mărimile de ambalaj să fie comercializate. Deţinătorul autorizaţiei de punere pe piaţă şi producătorul CSL Behring GmbH Emil-von-Behring-Strasse 76 D-35041 Marburg Germania Pentru orice informaţii despre acest medicament, vă rugăm să contactaţi reprezentantul local al deţinătorului autorizaţiei de punere pe piaţǎ: België/Belgique/Belgien CSL Behring NV Technologielaan 13 B-3001 Leuven Tél/Tel: +32 16 38 80 80
Luxembourg/Luxemburg CSL Behring NV Technologielaan 13 B-3001 Leuven Tél/Tel: +32 16 38 80 80
България CSL Behring GmbH Emil-von-Behring-Strasse 76 D-35041 Marburg Teл.: +49 64 213 93654
Magyarország Plazmed Kft. Fő u. 200 H-2193 Galgahéviz Tel.: +36 28 59 10 00

27
Česká republika IBP medica s.r.o. Pod Karlovem 8/1670 CZ 120 00 Praha 2 Tel: +42 02 22 56 07 23
Malta AM Mangion Ltd. Mangion Buildings New Street in Valletta Road MT- LQA 06 Luqa Tel: +356 2144 2010
Danmark CSL Behring ApS Lyngby Hovedgade 70B, 1.tv DK-2880 Kgs. Lyngby Tlf: +45 4520 1420
Nederland CSL Behring NV Claudius Prinsenlaan 128 NL-4818 CP Breda Tel: + 31 076 523 6045
Deutschland Wissenschaftliche Hotline CSL Behring GmbH Philipp-Reis-Str. 2 D-65795 Hattersheim Tel: +49 69 305 84437
Norge CSL Behring AB P.O.Box 712 S-182 17 Danderyd Tlf: +46 8 544 966 70
Eesti CSL Behring AB P.O.Box 712 S-182 17 Danderyd Tel: +46 8 544 966 70
Österreich CSL Behring GmbH Altmannsdorfer Straße 104 A-1121 Wien Tel: +43 1 80101 2463
Ελλάδα CSL Behring ΜΕΠΕ Mιχαλακοπούλου 35 GR-115 28 Αθήνα Τηλ: +30 210 7255 660
Polska Imed Poland sp. z.o.o. 3, Duchnicka ulica PL-01 796 Warszawa Tel.: +48 22 663 43 10
España CSL Behring S.A. Av Païssos Catalans 34 3º E-08950 Esplugues de Llobregat (Barcelona) Tel: +34 933 67 1870
Portugal CSL Behring Lta. Av 5 de Outubro, 198 – 3ºEsq. 1dto. P-1050-064 Lisboa Tel: +351 21 782 62 30
France CSL Behring 44 rue Cambronne F-75015 Paris Tél: + 33 1 53 58 54 00
România Prisum International Trading srl Strada Magura Vulturului 34 Sector 2 Bucureşti 021701 – RO Tel: +40 21 250 3688
Ireland CSL Behring Hayworth House, Market Place Haywards Heath RH16 1DB – UK Tel: +44 1444 447400
Slovenija MediSanus d.o.o. Vagajeva ulica 4 SI-1000 Ljubljana Tel: +386 1 518 33 97

28
Ísland CSL Behring AB P.O.Box 712 S-182 17 Danderyd Sími: +46 8 544 966 70
Slovenská republika TIMED, s.r.o. Trnavská cesta 112 SK-821 01 Bratislava Tel: +421 2 4820 95 11
Italia CSL Behring S.p.A. P.le Stefano Türr, 5 I-20149 Milano Tel: +39 02 34964 200
Suomi/Finland CSL Behring AB P.O.Box 712 S-182 17 Danderyd Puh/Tel: +46 8 544 966 70
Κύπρος ΑΚΗΣ ΠΑΝΑΓΙΩΤΟΥ & ΥΙΟΣ ΛΤΔ Γ. Κρανιδιώτη 4 CY-1522 Λευκωσία Τηλ: +357 22677038
Sverige CSL Behring AB P.O.Box 712 S-182 17 Danderyd Tel: +46 8 544 966 70
Latvija CSL Behring AB P.O.Box 712 S-182 17 Danderyd Tel: +46 8 544 966 70
United Kingdom CSL Behring Hayworth House, Market Place Haywards Heath RH16 1DB – UK Tel: +44 1444 447400
Lietuva CSL Behring AB P.O.Box 712 S-182 17 Danderyd Tel: +46 8 544 966 70
Acest prospect a fost aprobat în. Informaţii detaliate despre acest produs sunt disponibile pe website-ul Agenţiei Europene a Medicamentului (EMEA): http;//www.emea.europa.eu/