anexa i - ec.europa.eu · reykjavikurvegur 76-78 220 hafnarfjordur iceland ... via domenico...
TRANSCRIPT

1
Anexa I
Lista cu denumirile comerciale, formele farmaceutice, concentraţiile, căile de administrare ale medicamentelor, deţinătorii autorizaţiei de punere pe piaţă în statele membre
2
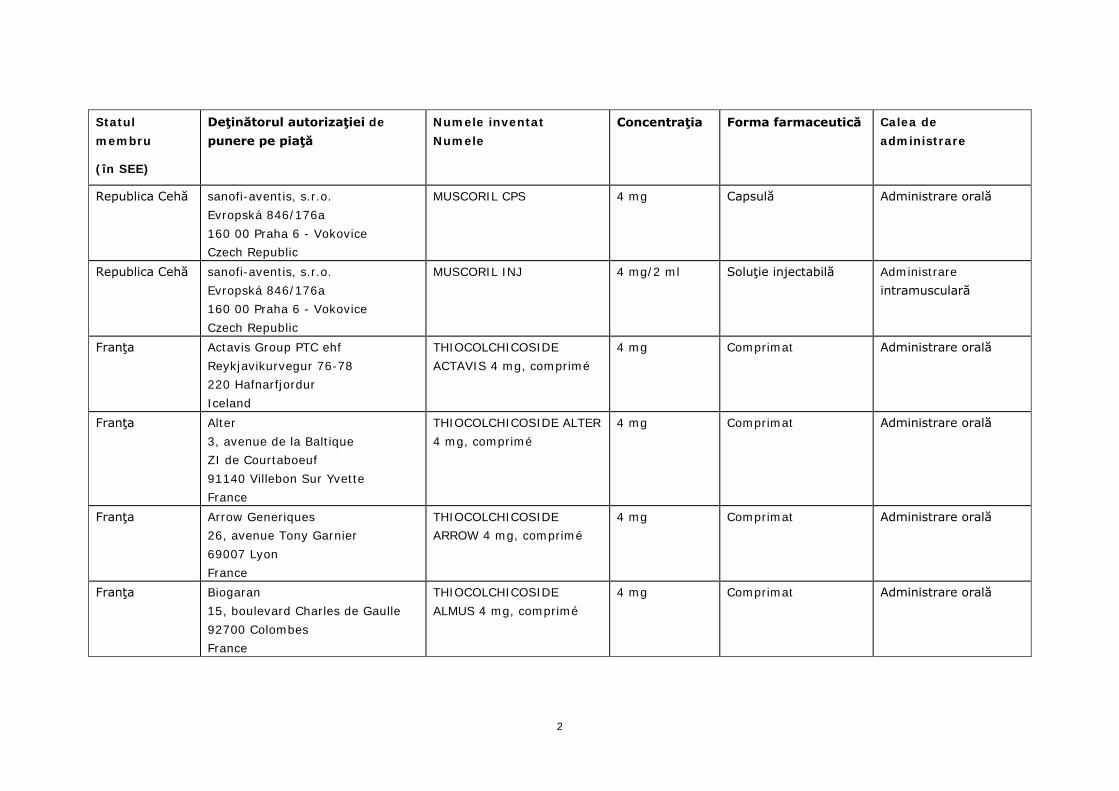
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
Republica Cehă sanofi-aventis, s.r.o. Evropská 846/176a 160 00 Praha 6 - Vokovice Czech Republic
MUSCORIL CPS 4 mg Capsulă Administrare orală
Republica Cehă sanofi-aventis, s.r.o. Evropská 846/176a 160 00 Praha 6 - Vokovice Czech Republic
MUSCORIL INJ 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Franţa Actavis Group PTC ehf Reykjavikurvegur 76-78 220 Hafnarfjordur Iceland
THIOCOLCHICOSIDE ACTAVIS 4 mg, comprimé
4 mg Comprimat Administrare orală
Franţa Alter 3, avenue de la Baltique ZI de Courtaboeuf 91140 Villebon Sur Yvette France
THIOCOLCHICOSIDE ALTER 4 mg, comprimé
4 mg Comprimat Administrare orală
Franţa Arrow Generiques 26, avenue Tony Garnier 69007 Lyon France
THIOCOLCHICOSIDE ARROW 4 mg, comprimé
4 mg Comprimat Administrare orală
Franţa Biogaran 15, boulevard Charles de Gaulle 92700 Colombes France
THIOCOLCHICOSIDE ALMUS 4 mg, comprimé
4 mg Comprimat Administrare orală
3
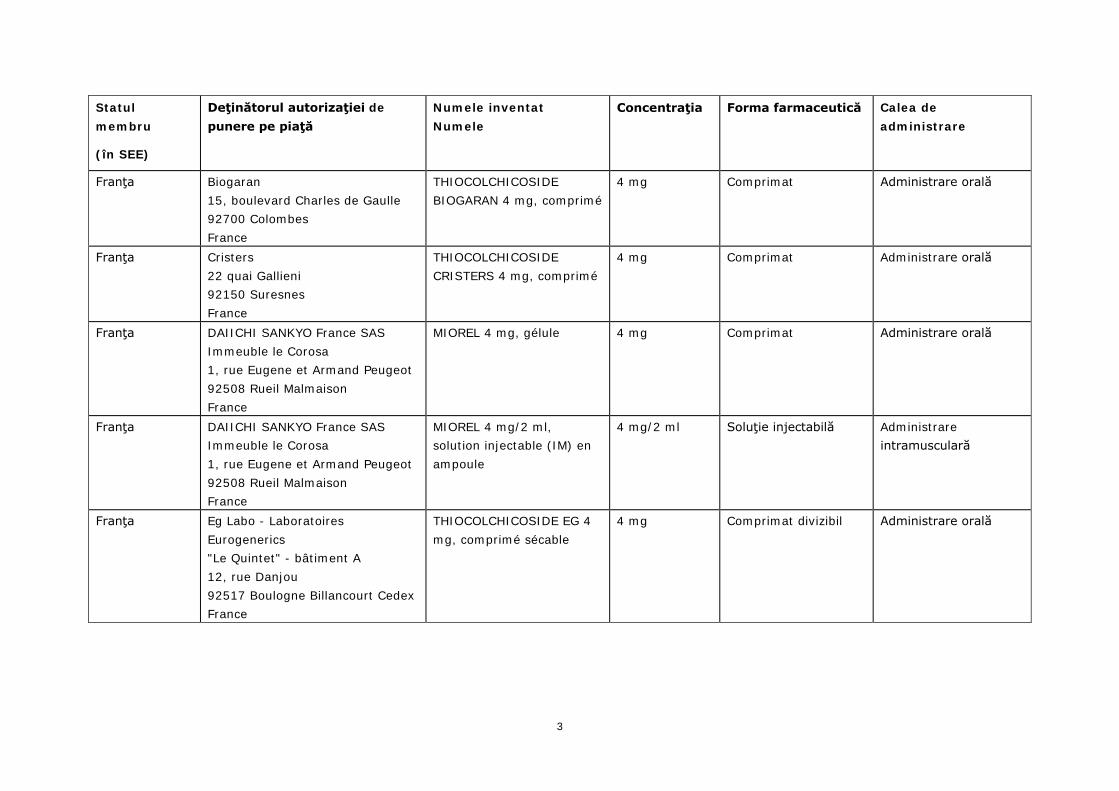
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
Franţa Biogaran 15, boulevard Charles de Gaulle 92700 Colombes France
THIOCOLCHICOSIDE BIOGARAN 4 mg, comprimé
4 mg Comprimat Administrare orală
Franţa Cristers 22 quai Gallieni 92150 Suresnes France
THIOCOLCHICOSIDE CRISTERS 4 mg, comprimé
4 mg Comprimat Administrare orală
Franţa DAIICHI SANKYO France SAS Immeuble le Corosa 1, rue Eugene et Armand Peugeot 92508 Rueil Malmaison France
MIOREL 4 mg, gélule 4 mg Comprimat Administrare orală
Franţa DAIICHI SANKYO France SAS Immeuble le Corosa 1, rue Eugene et Armand Peugeot 92508 Rueil Malmaison France
MIOREL 4 mg/2 ml, solution injectable (IM) en ampoule
4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Franţa Eg Labo - Laboratoires Eurogenerics "Le Quintet" - bâtiment A 12, rue Danjou 92517 Boulogne Billancourt Cedex France
THIOCOLCHICOSIDE EG 4 mg, comprimé sécable
4 mg Comprimat divizibil Administrare orală
4
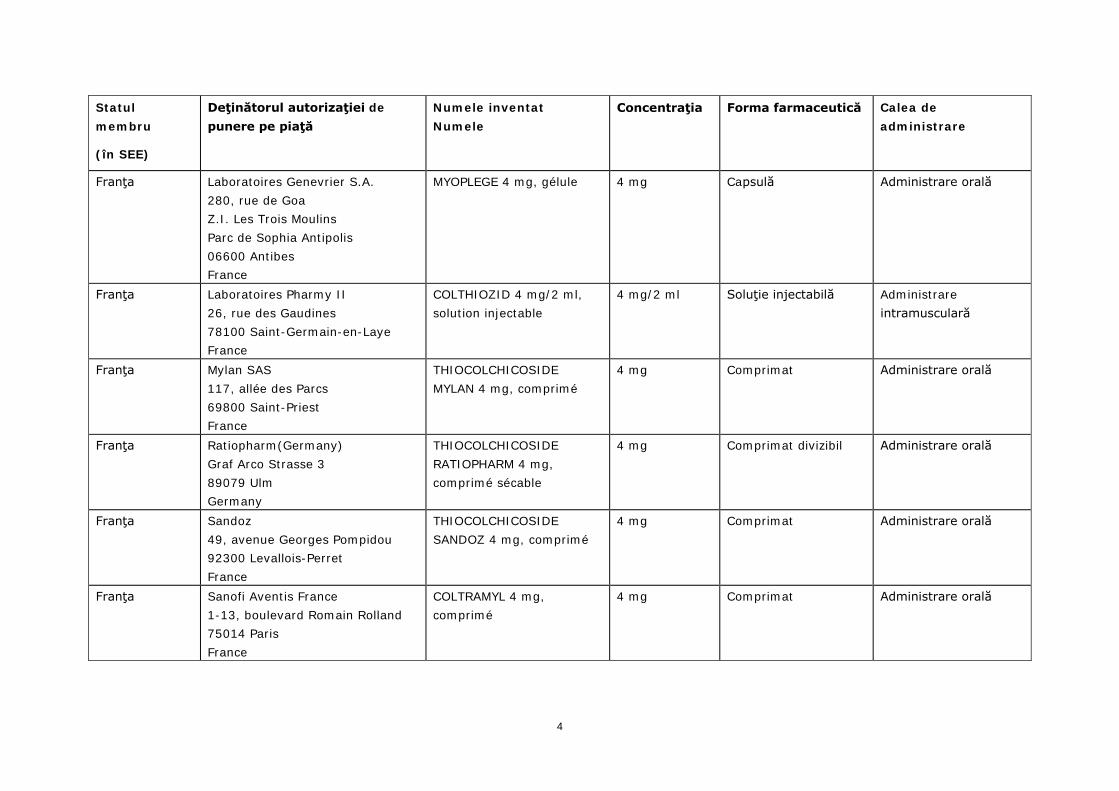
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
Franţa Laboratoires Genevrier S.A. 280, rue de Goa Z.I. Les Trois Moulins Parc de Sophia Antipolis 06600 Antibes France
MYOPLEGE 4 mg, gélule 4 mg Capsulă Administrare orală
Franţa Laboratoires Pharmy II 26, rue des Gaudines 78100 Saint-Germain-en-Laye France
COLTHIOZID 4 mg/2 ml, solution injectable
4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Franţa Mylan SAS 117, allée des Parcs 69800 Saint-Priest France
THIOCOLCHICOSIDE MYLAN 4 mg, comprimé
4 mg Comprimat Administrare orală
Franţa Ratiopharm(Germany) Graf Arco Strasse 3 89079 Ulm Germany
THIOCOLCHICOSIDE RATIOPHARM 4 mg, comprimé sécable
4 mg Comprimat divizibil Administrare orală
Franţa Sandoz 49, avenue Georges Pompidou 92300 Levallois-Perret France
THIOCOLCHICOSIDE SANDOZ 4 mg, comprimé
4 mg Comprimat Administrare orală
Franţa Sanofi Aventis France 1-13, boulevard Romain Rolland 75014 Paris France
COLTRAMYL 4 mg, comprimé
4 mg Comprimat Administrare orală
5
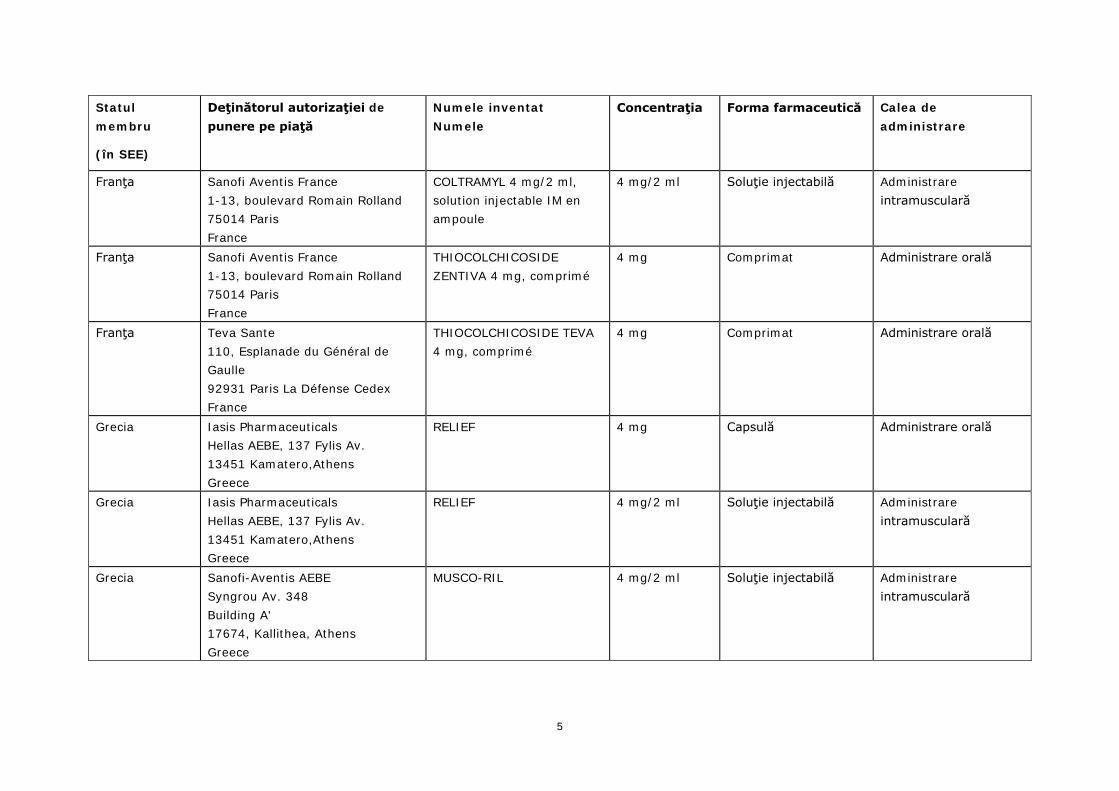
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
Franţa Sanofi Aventis France 1-13, boulevard Romain Rolland 75014 Paris France
COLTRAMYL 4 mg/2 ml, solution injectable IM en ampoule
4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Franţa Sanofi Aventis France 1-13, boulevard Romain Rolland 75014 Paris France
THIOCOLCHICOSIDE ZENTIVA 4 mg, comprimé
4 mg Comprimat Administrare orală
Franţa Teva Sante 110, Esplanade du Général de Gaulle 92931 Paris La Défense Cedex France
THIOCOLCHICOSIDE TEVA 4 mg, comprimé
4 mg Comprimat Administrare orală
Grecia Iasis Pharmaceuticals Hellas AEBE, 137 Fylis Av. 13451 Kamatero,Athens Greece
RELIEF 4 mg Capsulă Administrare orală
Grecia Iasis Pharmaceuticals Hellas AEBE, 137 Fylis Av. 13451 Kamatero,Athens Greece
RELIEF 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
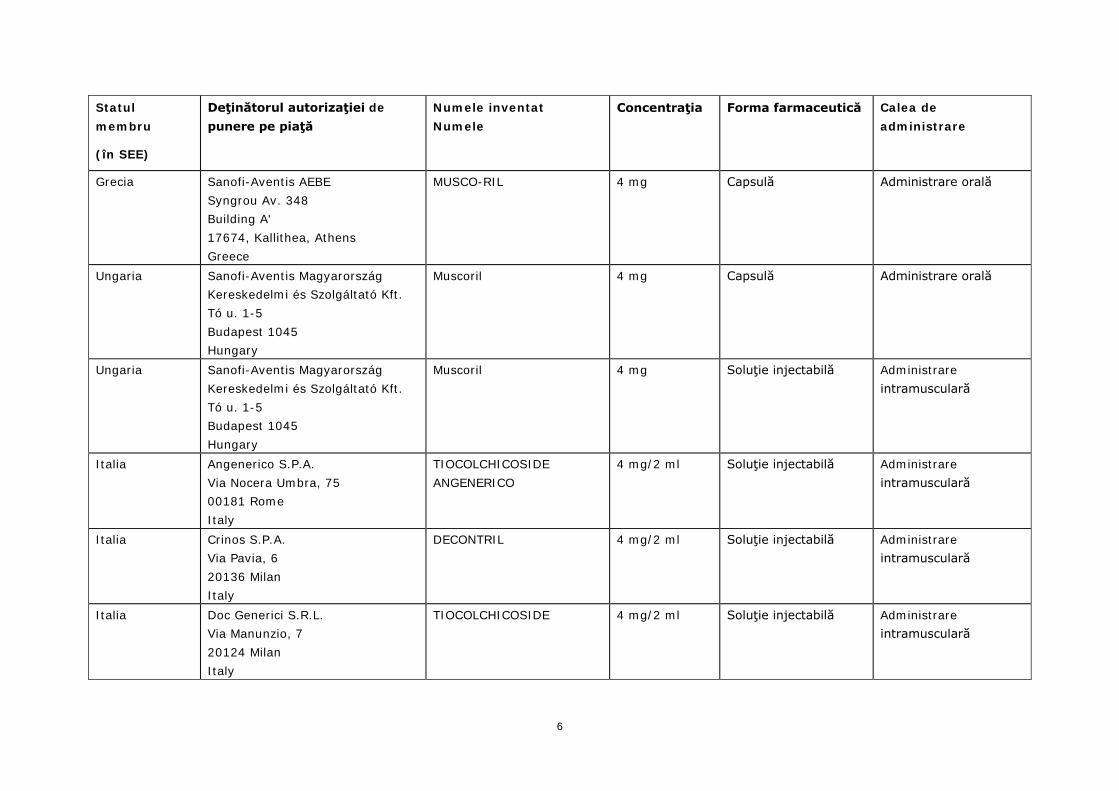
Grecia Sanofi-Aventis AEBE Syngrou Av. 348 Building A' 17674, Kallithea, Athens Greece
MUSCO-RIL 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
6
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
Grecia Sanofi-Aventis AEBE Syngrou Av. 348 Building A' 17674, Kallithea, Athens Greece
MUSCO-RIL 4 mg Capsulă Administrare orală
Ungaria Sanofi-Aventis Magyarország Kereskedelmi és Szolgáltató Kft. Tó u. 1-5 Budapest 1045 Hungary
Muscoril 4 mg Capsulă Administrare orală
Ungaria Sanofi-Aventis Magyarország Kereskedelmi és Szolgáltató Kft. Tó u. 1-5 Budapest 1045 Hungary
Muscoril 4 mg Soluţie injectabilă Administrare intramusculară
Italia Angenerico S.P.A. Via Nocera Umbra, 75 00181 Rome Italy
TIOCOLCHICOSIDE ANGENERICO
4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Crinos S.P.A. Via Pavia, 6 20136 Milan Italy
DECONTRIL 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Doc Generici S.R.L. Via Manunzio, 7 20124 Milan Italy
TIOCOLCHICOSIDE 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
7
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
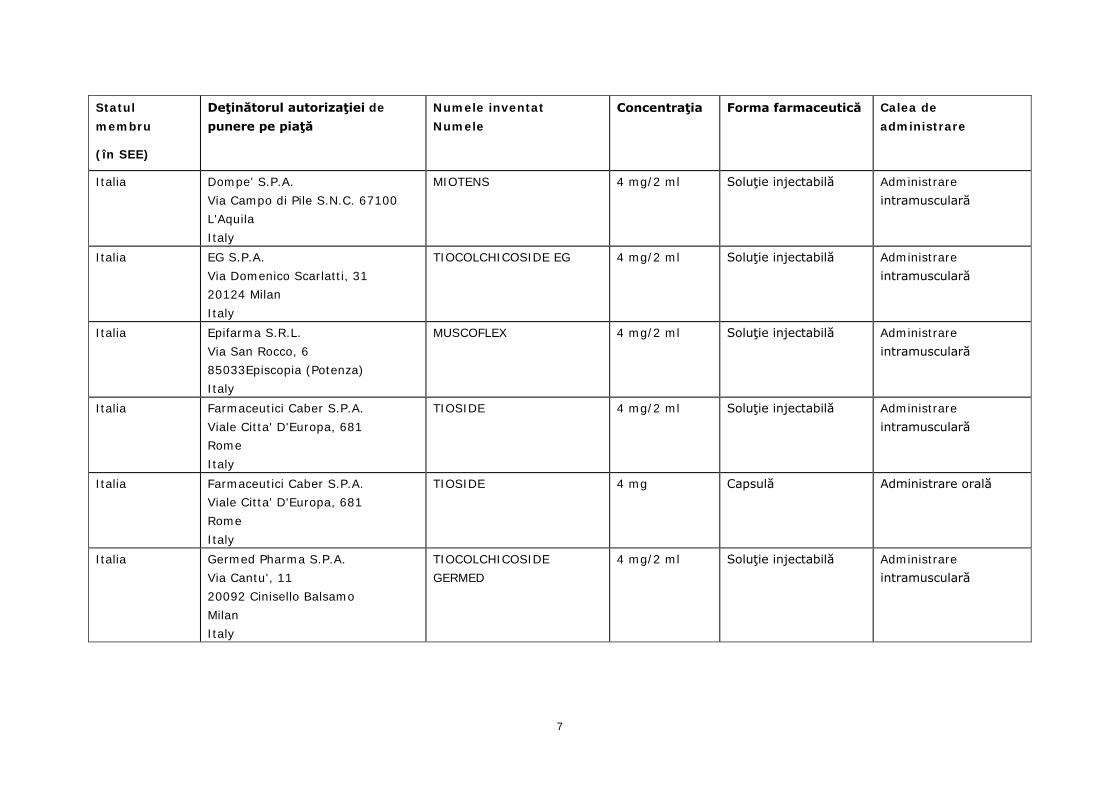
Italia Dompe' S.P.A. Via Campo di Pile S.N.C. 67100 L'Aquila Italy
MIOTENS 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia EG S.P.A. Via Domenico Scarlatti, 31 20124 Milan Italy
TIOCOLCHICOSIDE EG 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Epifarma S.R.L. Via San Rocco, 6 85033Episcopia (Potenza) Italy
MUSCOFLEX 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Farmaceutici Caber S.P.A. Viale Citta' D'Europa, 681 Rome Italy
TIOSIDE 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Farmaceutici Caber S.P.A. Viale Citta' D'Europa, 681 Rome Italy
TIOSIDE 4 mg Capsulă Administrare orală
Italia Germed Pharma S.P.A. Via Cantu', 11 20092 Cinisello Balsamo Milan Italy
TIOCOLCHICOSIDE GERMED
4 mg/2 ml Soluţie injectabilă Administrare intramusculară
8
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
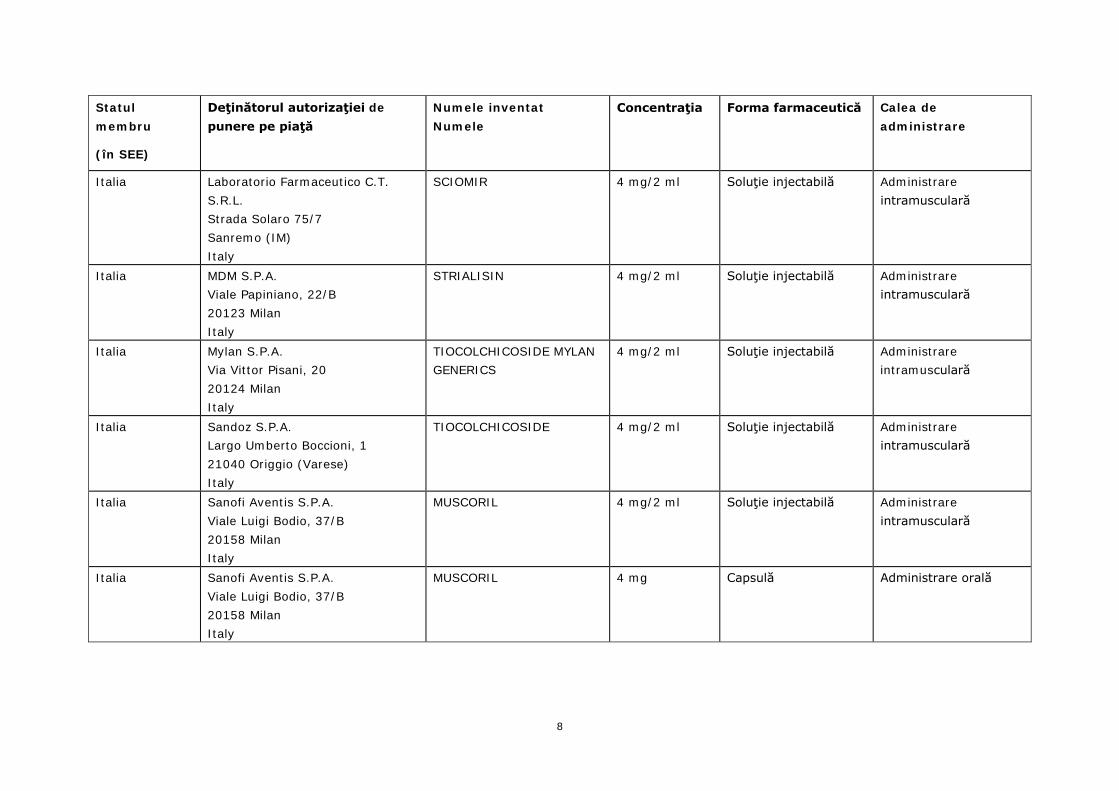
Italia Laboratorio Farmaceutico C.T. S.R.L. Strada Solaro 75/7 Sanremo (IM) Italy
SCIOMIR 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia MDM S.P.A. Viale Papiniano, 22/B 20123 Milan Italy
STRIALISIN 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Mylan S.P.A. Via Vittor Pisani, 20 20124 Milan Italy
TIOCOLCHICOSIDE MYLAN GENERICS
4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Sandoz S.P.A. Largo Umberto Boccioni, 1 21040 Origgio (Varese) Italy
TIOCOLCHICOSIDE 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Sanofi Aventis S.P.A. Viale Luigi Bodio, 37/B 20158 Milan Italy
MUSCORIL 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Sanofi Aventis S.P.A. Viale Luigi Bodio, 37/B 20158 Milan Italy
MUSCORIL 4 mg Capsulă Administrare orală
9
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
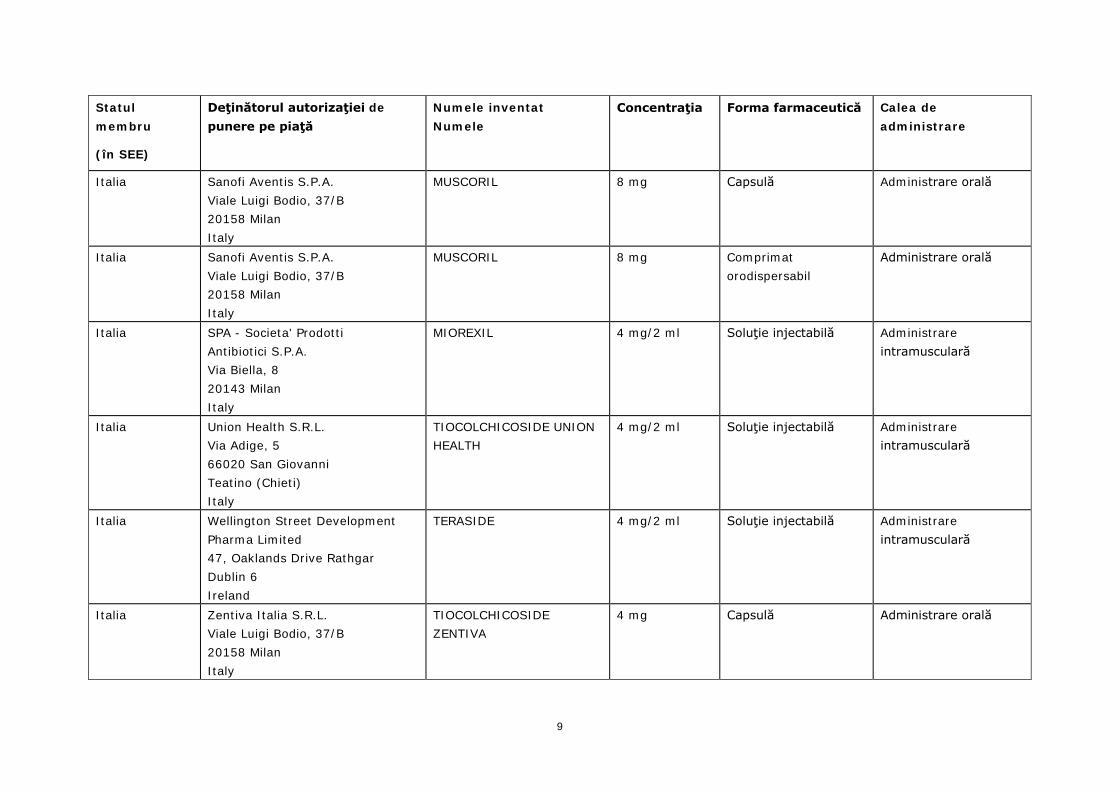
Italia Sanofi Aventis S.P.A. Viale Luigi Bodio, 37/B 20158 Milan Italy
MUSCORIL 8 mg Capsulă Administrare orală
Italia Sanofi Aventis S.P.A. Viale Luigi Bodio, 37/B 20158 Milan Italy
MUSCORIL 8 mg Comprimat orodispersabil
Administrare orală
Italia SPA - Societa' Prodotti Antibiotici S.P.A. Via Biella, 8 20143 Milan Italy
MIOREXIL 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Union Health S.R.L. Via Adige, 5 66020 San Giovanni Teatino (Chieti) Italy
TIOCOLCHICOSIDE UNION HEALTH
4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Italia Wellington Street Development Pharma Limited 47, Oaklands Drive Rathgar Dublin 6 Ireland
TERASIDE 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
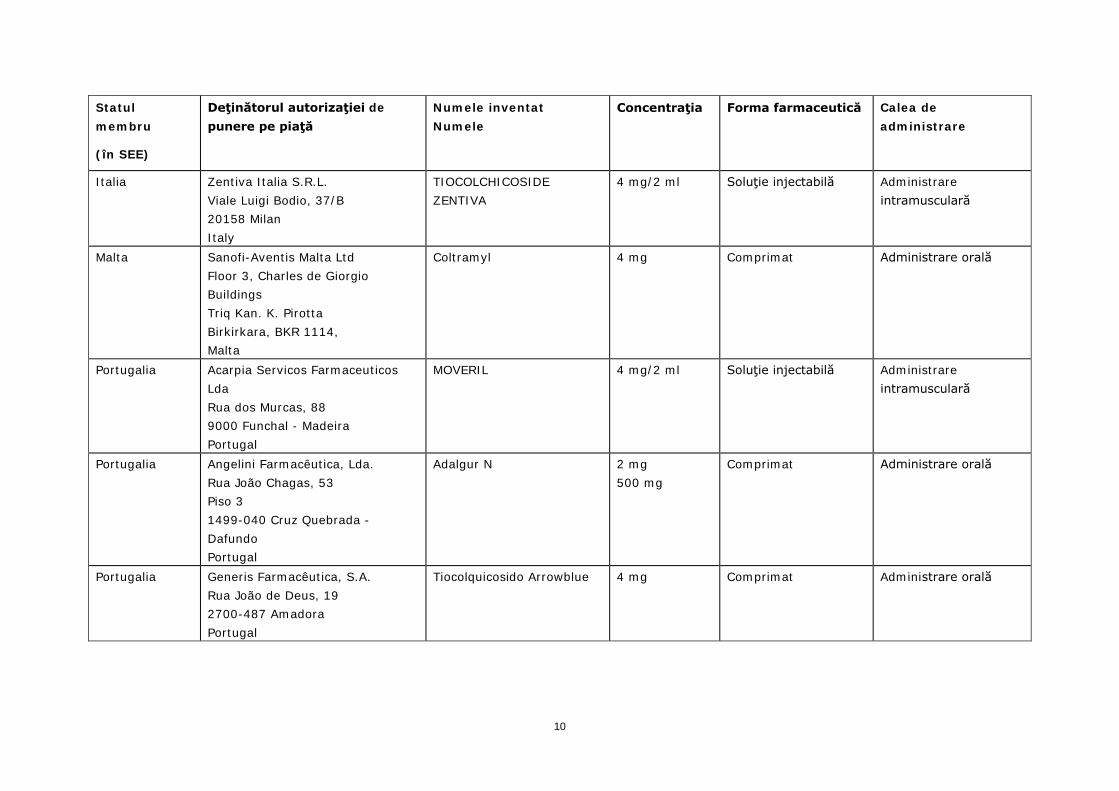
Italia Zentiva Italia S.R.L. Viale Luigi Bodio, 37/B 20158 Milan Italy
TIOCOLCHICOSIDE ZENTIVA
4 mg Capsulă Administrare orală
10
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
Italia Zentiva Italia S.R.L. Viale Luigi Bodio, 37/B 20158 Milan Italy
TIOCOLCHICOSIDE ZENTIVA
4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Malta Sanofi-Aventis Malta Ltd Floor 3, Charles de Giorgio Buildings Triq Kan. K. Pirotta Birkirkara, BKR 1114, Malta
Coltramyl 4 mg Comprimat Administrare orală
Portugalia Acarpia Servicos Farmaceuticos Lda Rua dos Murcas, 88 9000 Funchal - Madeira Portugal
MOVERIL 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Portugalia Angelini Farmacêutica, Lda. Rua João Chagas, 53 Piso 3 1499-040 Cruz Quebrada - Dafundo Portugal
Adalgur N 2 mg 500 mg
Comprimat Administrare orală
Portugalia Generis Farmacêutica, S.A. Rua João de Deus, 19 2700-487 Amadora Portugal
Tiocolquicosido Arrowblue 4 mg Comprimat Administrare orală
11
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
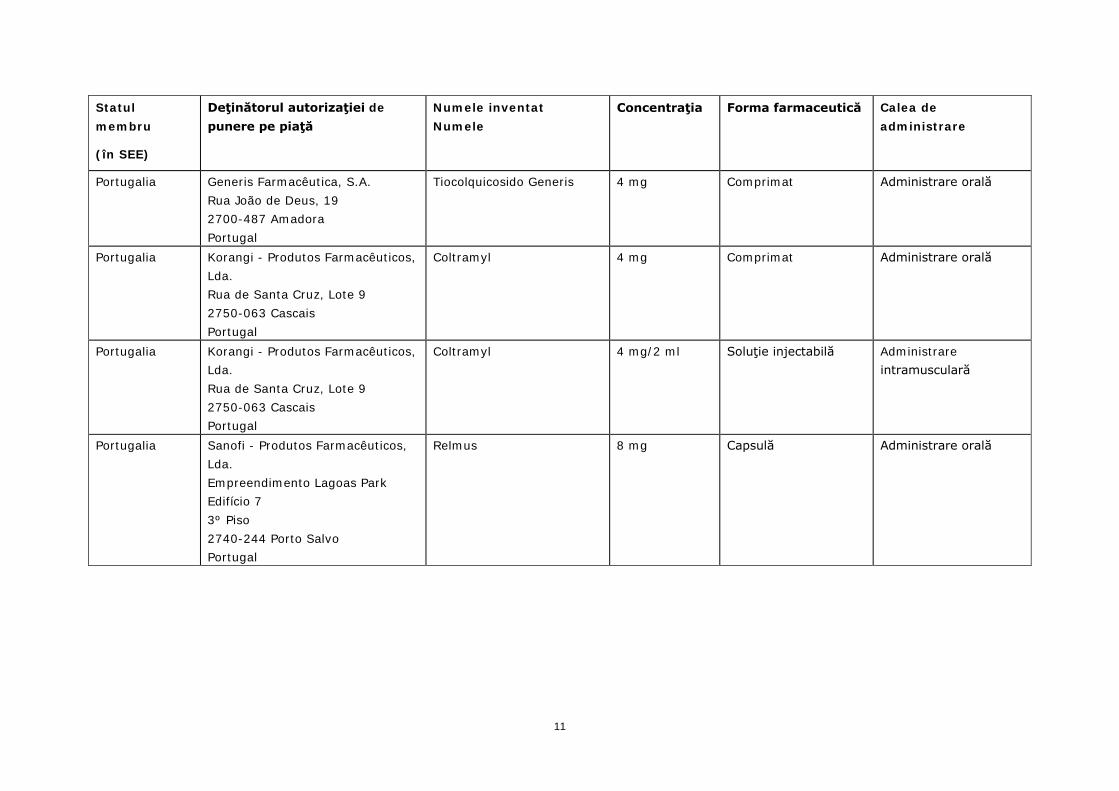
Portugalia Generis Farmacêutica, S.A. Rua João de Deus, 19 2700-487 Amadora Portugal
Tiocolquicosido Generis 4 mg Comprimat Administrare orală
Portugalia Korangi - Produtos Farmacêuticos, Lda. Rua de Santa Cruz, Lote 9 2750-063 Cascais Portugal
Coltramyl 4 mg Comprimat Administrare orală
Portugalia Korangi - Produtos Farmacêuticos, Lda. Rua de Santa Cruz, Lote 9 2750-063 Cascais Portugal
Coltramyl 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Portugalia Sanofi - Produtos Farmacêuticos, Lda. Empreendimento Lagoas Park Edifício 7 3º Piso 2740-244 Porto Salvo Portugal
Relmus 8 mg Capsulă Administrare orală
12
Statul membru
(în SEE)
Deţinătorul autorizaţiei de punere pe piaţă
Numele inventat Numele
Concentraţia Forma farmaceutică Calea de administrare
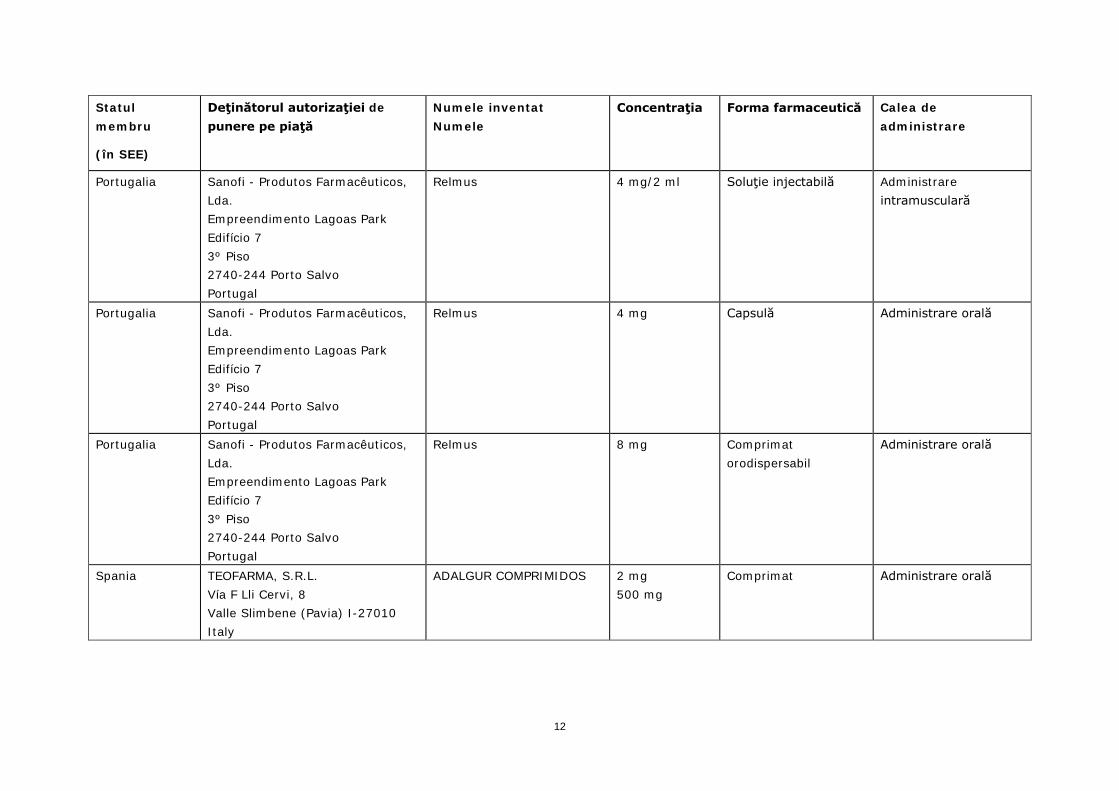
Portugalia Sanofi - Produtos Farmacêuticos, Lda. Empreendimento Lagoas Park Edifício 7 3º Piso 2740-244 Porto Salvo Portugal
Relmus 4 mg/2 ml Soluţie injectabilă Administrare intramusculară
Portugalia Sanofi - Produtos Farmacêuticos, Lda. Empreendimento Lagoas Park Edifício 7 3º Piso 2740-244 Porto Salvo Portugal
Relmus 4 mg Capsulă Administrare orală
Portugalia Sanofi - Produtos Farmacêuticos, Lda. Empreendimento Lagoas Park Edifício 7 3º Piso 2740-244 Porto Salvo Portugal
Relmus 8 mg Comprimat orodispersabil
Administrare orală
Spania TEOFARMA, S.R.L. Vía F Lli Cervi, 8 Valle Slimbene (Pavia) I-27010 Italy
ADALGUR COMPRIMIDOS 2 mg 500 mg
Comprimat Administrare orală

13
Anexa II
Concluzii ştiinţifice şi motivele modificării termenilor autorizaţiilor de punere pe piaţă

14
Concluzii ştiinţifice Rezumat general al evaluării ştiinţifice pentru medicamentele care conţin tiocolchicozidă pentru uz sistemic (vezi anexa I) Tiocolchicozida (TCC) este un derivat semisintetic sulfurat al colchicozidei, cu activitate farmacologică de miorelaxare. Administrarea miorelaxantelor reprezintă unul dintre numeroasele tratamente utilizate în prezent în gestionarea terapeutică a durerilor lombare nespecifice. Utilizarea TCC este indicată în tratamentul contracturilor musculare dureroase cu diverse localizări. Beneficiile medicamentelor care conţin TCC sunt recunoscute în practica clinică, fiind utilizate la scară largă de medicii prescriptori din statele membre interesate (vezi anexa I). În urma întreruperii unui studiu clinic de fază I cu TCC de către o companie, datorită noilor rezultate nonclinice, agenţia italiană de reglementare în domeniul medicamentelor (AIFA) a solicitat unuia dintre deţinătorii autorizaţiilor de punere pe piaţă (DAPP) pentru TCC să investigheze potenţialul genotoxic al TCC şi, în special, al metaboliţilor acesteia; deţinătorului autorizaţiei de punere pe piaţă i s-a solicitat să efectueze studii preclinice in vivo şi in vitro în care să evalueze genotoxicitatea potenţială a metaboliţilor tiocolchicozidei. Rezultatele obţinute din unul dintre studiile care au analizat metaboliţii (metabolitul SL59.0955, M2) au determinat motive de îngrijorare: noile date obţinute din studiile preclinice cu privire la efectul aneugenic al metabolitului M2 al TCC au indicat un semnal de potenţial genotoxic. Având în vedere cele menţionate mai sus, la 15 februarie 2013, Italia a solicitat CHMP, în temeiul articolului 31 din Directiva 2001/83/CE, să evalueze motivele de îngrijorare formulate mai sus privind aneuploidia şi impactul acesteia asupra raportului beneficiu-risc pentru medicamentele care conţin TCC pentru uz sistemic. A fost solicitat avizul CHMP privind restricţionarea indicaţiei pentru medicamentele care conţin TCC şi/sau luarea altor măsuri de reglementare. La 21 februarie 2013, CHMP a iniţiat o procedură de sesizare pentru medicamentele care conţin TCC. Aneuploidia (modificarea numărului de cromozomi şi pierderea heterozigozităţii) este recunoscută ca fiind un factor de risc cu potenţial cancerigen atunci când are impact asupra celulelor somatice şi un factor de risc de teratogenitate, embriotoxicitate/avorturi spontane şi afectarea fertilităţii masculine când are impact asupra celulelor embrionare1. În scopul evaluării acestor riscuri, deţinătorii autorizaţiilor de punere pe piaţă au prezentat o analiză a potenţialului genotoxic pentru fiecare cale de administrare sistemică, împreună cu o analiză a posibililor factori de risc, inclusiv criteriile relevante, cum ar fi doza şi durata tratamentului. CHMP a evaluat toate datele disponibile din studiile preclinice şi clinice, din literatura de specialitate şi din experienţa ulterioară punerii pe piaţă privind aneuploidia în cazul medicamentelor care conţin TCC pentru uz sistemic. Este prezentat în continuare un rezumat care vizează aceste aspecte. Studii preclinice
Dezvoltarea preclinică a TCC s-a realizat în principal în anii 1980 şi a fost completată în anii 1990 pentru a fi în conformitate cu „European guidelines on the non-clinical documentation for mixed marketing authorisation applications” (Ghidurile europene privind documentaţia non-clinică pentru cererile mixte de acordare a autorizaţiei de introducere pe piaţă) (CPMP/SWP/799/95) şi pentru a investiga un metabolit activ nou, SL18.0740 (M1), care a fost identificat în acea perioadă. În anii 20012 şi 20033 au fost publicate evaluări ulterioare privind siguranţa, care s-au concentrat asupra potenţialului genotoxic. După sistarea studiului clinic de fază I referitor la TCC menţionat mai sus, potenţialul genotoxic al metabolitului aglicon SL59.0955 (M2) a continuat să fie investigat. Au fost efectuate studii noi în anii 2011 şi 2012 referitoare la genotoxicitatea substanţei active nemodificate (TCC), a principalului său metabolit circulant SL18.0740 şi a metabolitului aglicon SL59.0955. Date de genotoxicitate referitoare la TCC şi la principalul său metabolit circulant SL18.0740 (M1) Au fost efectuate diverse studii de toxicologie genetică referitoare la TCC şi la principalul său metabolit identificat 3-O-glucuronidat aglicon (SL18.0740), care este metabolitul activ.
1 Parry 2000 & 2002; Kirsch-Volders 2002 2 Kirkland DJ Et al. 2001 3 Gouy D., 2003

15
S-a concluzionat că M1 (SL18.0740) este lipsit de potenţial mutagen (mutaţii genice) şi clastogen (leziuni cromozomiale structurale), însă este capabil să inducă aneuploidia (modificarea numărului de cromozomi). Cu toate acestea, într-un studiu de urmărire (testul in vivo pe micronuclei) a fost definit un nivel la care nu s-a observat niciun efect, la 39,6 mg/kg. Acest fapt a fost asociat cu o ASC plasmatică a lui M1 egală cu 4073 ng.h/ml, care este de 20 de ori mai mare decât expunerea la M1 observată la om după administrarea pe cale orală a unei doze de 8 mg de TCC de două ori pe zi (175 ng.h/ml la 30 de minute). Prin urmare, pe baza datelor disponibile menţionate mai sus, CHMP a considerat că limitele de siguranţă şi raportul beneficiu-risc pentru TCC şi SL18.0740 (M1) sunt acceptabile. Date de genotoxicitate referitoare la metabolitul aglicon SL59.0955 (M2) Întrucât nu a fost efectuat anterior niciun studiu relevant de toxicologie genetică cu metabolitul aglicon SL59.0955, au fost desfăşurate studii complementare (teste ale leziunilor cromozomiale) în scopul investigării profilului genotoxic al acestui metabolit şi al capacităţii sale de a induce aneuploidia, prin teste non-clinice in vitro (până la 600 µg/ml) şi in vivo (până la 150 µg/ml):
• un test in vitro al micronucleilor (MN) prin utilizarea unei culturi primare de limfocite umane cu metabolitul aglicon (SL59.0955), prin colorare centromerică (Whitwell J., 2012);
• un test in vivo al MN din măduva osoasă de şobolan, în urma administrării metabolitului aglicon (SL59.0955) pe cale orală la şobolani, prin colorare centromerică şi printr-o evaluare completă a expunerii la SL59.0955 şi la metabolitul 3-O-glucuronidat aglicon (SL18.0740) pentru o evaluare mai bună a pragului de expunere (Wase K., octombrie 2012).
Testul in vitro al MN din limfocitele umane a arătat că M2 a determinat inducţia micronucleară pe culturile de limfocite umane din sângele periferic în toate condiţiile de tratament. Analiza mecanistică ulterioară prin utilizarea hibridizării in situ cu fluorescenţă (HISF) cu sonde ADN pancentromerice a demonstrat că micronucleii au fost generaţi în mod predominant prin intermediul unui mecanism aneugen (număr anormal de cromozomi) în toate condiţiile de tratament; aneuploidia a fost confirmată în mod evident prin metoda de colorare centromerică. În condiţiile testului, nivelul la care nu s-au observat efecte adverse (NOAEL) şi nivelul cel mai scăzut la care au fost observate efecte adverse (LOAEL) au fost, de asemenea, avute în vedere, însă, deşi a fost recunoscut faptul că non-disjuncţia cromozomială (NDC) este criteriul final cel mai adecvat care trebuie investigat pentru determinarea efectelor toxinelor fusocelulare în doză mică, nu a fost posibil să se ajungă la o concluzie privind identificarea unui prag al dozelor pentru inducţia aneuploidiei. În testul in vivo al MN din măduva osoasă de şobolan, în urma administrării pe cale orală a M2 o dată pe zi, timp de două zile consecutive, la doze de 25, 50, 70, 100 sau 150 mg/kg/zi, testul micronucleilor din măduva osoasă la masculii de şobolan a dat rezultat negativ. La femele, a fost observat un răspuns pozitiv la doze de 25, 50, 70 şi 100 mg/kg/zi, pe baza mediei grupului analizat şi pe baza datelor individuale. Este cunoscut faptul că mecanismele genotoxice, precum aneuploidia, care implică diviziunea celulară şi segmente ţintă, altele decât ADN, apar peste un anumit prag de expunere. Cu toate acestea, în cazul efectelor aneugenice, la femelele de şobolan nu a fost identificat niciun nivel la care nu s-au observat efecte adverse (LOEL = 25 mg/kg) şi nu s-a observat niciun efect evident asociat dozei, deoarece a fost remarcată doar o uşoară diferenţă de expunere (ASC0-24 şi Cmax) cu 3-demetiltiocolchicină (SL59.0955) între dozele diferite administrate la masculi şi femele. În plus, la masculi şi femele s-a observat doar o diferenţă minoră de expunere legată de sex. Prin urmare, nu a putut fi calculată nicio limită de siguranţă. Efectul aneugenic a fost observat la nivelul LOEL care a corespuns cu numai 1,6 x Cmax umană şi cu 4,1 x ASC [8 mg de două ori pe zi, Per Os (PO)]. După administrarea pe cale parenterală, se preconizează o concentraţie plasmatică a M2 mult mai mică, deoarece metabolizarea M2 se produce după administrarea pe cale orală, în principal prin metabolizare la nivelul intestinului. Cu toate acestea, nu se ştie dacă expunerea la M2 s-ar putea situa sub pragul de aneugenitate (care cuprinde o limită de siguranţă suficient de mare), deoarece M2 nu a fost analizat în studiile clinice cinetice disponibile. În concluzie, rezultatele din studiile pre-clinice menţionate mai sus au arătat că M2 (SL59.0955) a determinat inducţia micronucleară in vitro şi in vivo, micronucleii fiind produşi în mod predominant prin intermediul mecanismului aneugen în toate condiţiile de tratament. În cadrul celor două studii

16
pre-clinice in vitro şi in vivo, rezultatele (creşterea incidenţei celulelor micronucleate) au fost observate la concentraţii/expuneri apropiate de expunerile măsurate la om, la dozele terapeutice. Prin urmare, în opinia CHMP, datele disponibile permit confirmarea unui efect aneugenic evident al metabolitului tiocolchicozidă M2 la concentraţii care sunt de 4 ori mai mari decât expunerea plasmatică la om după un tratament cu TCC în doză orală de 8 mg de două ori pe zi (doza recomandată) şi cu o doză iniţială de 25 mg/kg. Datele prezentate nu au permis stabilirea unui nivel NOEL pentru aneuploidie şi, prin urmare, nu este exclus un risc potenţial la om . Siguranţă clinică
Deţinătorii autorizaţiilor de punere pe piaţă au depus studii clinice şi rapoarte spontane ulterioare punerii pe piaţă. Studii clinice Nu au fost identificate cazuri de cancer, malformaţii congenitale, avort spontan şi afectare a fertilităţii masculine în evaluarea studiilor clinice şi în literatura de specialitate. Experienţa ulterioară punerii pe piaţă Cazurile spontane ulterioare punerii pe piaţă au fost colectate pe baza rapoartelor înregistrate în două baze de date generale de farmacovigilenţă ale deţinătorilor autorizaţiilor de punere pe piaţă (datelimită: 15 februarie 2013 şi, respectiv, 29 aprilie 2013) În prima bază de date, au fost raportate 11 cazuri secundare expunerii din timpul sarcinii:
• şase cazuri de malformaţii congenitale (adică un caz de malformaţii multiple care au dus la avort, un caz de hipoplazie pulmonară, un caz de palatoschizis, un caz de spina bifida, un caz de sindrom Poland, un caz de persistenţă a canalului arterial),
• patru cazuri de avort spontan, • un caz de iminenţă de naştere prematură.
Rapoartele de evaluare a cazurilor din a doua bază de date, înregistrate pe perioada situată între anul 2004 şi până pe 29 aprilie 2013, au raportat 23 de cazuri secundare expunerii în timpul sarcinii şi/sau al expunerii în timpul dezvoltării intrauterine:
• 20 de cazuri provocate de expunerea pe parcursul perioadei de dezvoltare embrionară, dintre care:
o două cazuri de efecte (malformaţii) teratogene asociate cu expunerea la începutul sarcinii (primul trimestru este perioada în care riscul este cel mai semnificativ),
o patru cazuri care au avut ca rezultat întreruperea sarcinii (3 avorturi spontane şi un avort indus, care nu s-a datorat unui motiv medical)
o cinci cazuri cu evoluţie favorabilă (fără efect asupra nou-născutului) o nouă cazuri cu evoluţie necunoscută a sarcinii din cauza lipsei de documentaţie.
• 1 caz provocat de expunerea în perioada de dezvoltare fetală [adică un caz de efecte feto-toxice care a avut ca rezultat un tip de afectare fetală sau neonatală cu impact asupra realizării creşterii sau a maturizării histologice sau funcţionale a organelor în perioada de dezvoltare a acestora (perioada în care apar riscurile cele mai mari din al doilea trimestru de sarcină)],
• şi 2 cazuri provocate de expunerea într-o perioadă necunoscută a sarcinii: o 1 caz de efecte teratogene (malformaţii) asociate cu expunerea la începutul
sarcinii, o 1 caz cu evoluţie necunoscută a sarcinii din cauza lipsei de documentaţie.
Nu a fost înregistrat niciun caz de efecte neonatale legate de expunere în ultima perioadă a sarcinii sau în cursul naşterii. CHMP consideră că dovezile clinice din ansamblul de cazuri raportate de deţinătorii autorizaţiilor de punere pe piaţă cu privire la consecinţele aneuploidiei la om nu permit stabilirea unor concluzii definitive. Aneuploidia este o caracteristică a celulelor canceroase observată frecvent. Cu toate

17
acestea, dacă aneuploidia are o contribuţie cauzală sau este pur şi simplu o consecinţă a transformărilor neoplazice continuă să fie un subiect controversat. În plus, lipsa de dovezi privind corelaţia dintre utilizarea TCC şi cancer s-ar putea datora dificultăţii în stabilirea unei relaţii de cauzalitate între administrarea medicamentului şi efectul acestuia, care poate să apară după mai mulţi ani de la utilizare. În majoritatea cazurilor, tratamentul este pentru utilizare pe termen scurt şi nu este asociat cu percepţia nici de către medici, nici de către pacienţi, a unui risc crescut de cancer, prin urmare este dificil să se stabilească o relaţie de cauzalitate între apariţia cancerului şi tratament. De asemenea, CHMP a observat că numărul limitat de cazuri de malformaţii/toxicitate embrio-fetală se poate datora faptului că, în majoritatea statelor membre, administrarea medicamentului este contraindicată în timpul sarcinii. Ţinând cont de totalitatea datelor, CHMP a considerat că relaţia de cauzalitate nu poate fi exclusă şi că aneuploidia trebuie considerată teoretic ca fiind un factor de risc pentru apariţia cancerului. Prin urmare, CHMP a fost de părere că trebuie luate măsuri de reducere la minimum a riscurilor (MRMR) în scopul abordării riscurilor de teratogenitate, embriotoxicitate/avorturi spontane, afectarea fertilităţii masculine şi cancer.
• În primul rând, întrucât s-a demonstrat că metabolitul M2 al TCC este aneugen la niveluri de expunere apropiate celor de expunere terapeutică la om, CHMP a considerat că doza trebuie restricţionată (la 8 mg de două ori pe zi PO şi la 4 mg de două ori pe zi pe cale intramusculară) şi că trebuie evitată utilizarea pe termen lung. În această privinţă, CHMP a fost de părere că indicaţia în „boala Parkinson şi simptome parkinsoniene induse medicamentos, cu atenţie deosebită la sindromul dislexiei neurologice” trebuie eliminată, deoarece aceasta este o indicaţie pentru utilizare cronică. De asemenea, CHMP a considerat că utilizarea TCC trebuie evitată în timpul pubertăţii (12 până la 16-18 ani) din cauza potenţialului risc asupra fertilităţii. Aşadar, utilizarea produsului trebuie să se limiteze la afecţiunile acute manifestate de pacienţii cu vârsta de peste 16 ani; în consecinţă, a fost aprobat un Rezumat al caracteristicilor produsului (RCP) cu restricţii pentru utilizarea şi durata terapiei. Pe baza utilizării frecvente în cadrul afecţiunilor acute, au fost incluse alte recomandări privind dozele pentru limitarea duratei tratamentului la 7 zile în cazul administrării pe cale orală şi la 5 zile în cazul administrării intramusculare; de asemenea, a fost recomandată referirea la doza maximă permisă. În final, a fost solicitat un interval de 12 ore între două administrări consecutive, din cauza timpului de înjumătăţire prin eliminare al metabolitului M2. Punctele corespunzătoare din informaţiile referitoare la produs au fost actualizate în consecinţă. În plus, CHMP a fost de părere că mărimea ambalajului trebuie limitată în funcţie de noua schemă de tratament recomandată, în funcţie de numărul de zile (ambalaj cu maximum 30 de comprimate sau capsule/4 mg sau ambalaj cu maximum 14 comprimate sau capsule/8 mg şi ambalaj cu maximum 10 flacoane/fiole).
• Teratogenitatea este clasificată ca fiind un important risc identificat. Pentru a aborda
riscurile de teratogenitate şi embriotoxicitate/avorturi spontane, CHMP a fost de acord ca TCC să fie contraindicată pe parcursul întregii perioade de sarcină, în timpul lactaţiei şi la femeile cu potenţial fertil care nu utilizează medicamente contraceptive. De asemenea, au fost aprobate modificările de la punctele privind atenţionările şi perioada de sarcină şi lactaţie din informaţiile referitoare la produs.
• Carcinogenitatea şi afectarea fertilităţii sunt clasificate ca fiind importante riscuri potenţiale. Referitor la riscul de infertilitate masculină, se ştie că aneuploidia caracterizată printr-un număr crescut de cromozomi în spermă este asociată cu infertilitatea masculină. Cu toate acestea, au fost ridicate mai multe motive de îngrijorare în ceea ce priveşte potenţialul risc de anomalii fetale din cauza aneuploidiei caracterizate prin număr crescut de spermatozoizi decât în ceea ce priveşte infertilitatea masculină în sine. Date fiind condiţiile de tratament cu TCC (pe termen scurt, cu potenţial aneugenic la doze maxime), efectele asupra fertilităţii masculine vor fi reduse şi poate fi preconizată o revenire rapidă la nivelurile normale. Pentru a aborda acest motiv de îngrijorare, a fost aprobată o modificare în informaţiile referitoare la produs.
• În sfârşit, dovezile de carcinogenitate a medicamentelor aneugenice sunt limitate. În
general, riscul de cancer crescut în mod semnificativ ar depinde de expunerea/dozarea pe

18
termen lung/cronică a medicamentului aneugenic. Carcinogenitatea este un important risc potenţial. În abordarea acestuia, CHMP a considerat că măsurile de reducere la minimum a riscurilor (MRMR) (limitarea indicaţiei la afecţiunile acute, limitarea duratei de tratament la şapte zile consecutive, evitarea utilizării pe termen lung) au fost adecvate.
CHMP a considerat că era necesară o Comunicare directă către profesioniştii din domeniul sănătăţii (DHPC) pentru a informa cu privire la rezultatul acestei evaluări, inclusiv cu privire la indicaţia actualizată, utilizarea clinică pentru aceste produse (pe termen scurt) şi pentru a evidenţia riscul genotoxic. Un plan de management al riscurilor (PMR) va fi transmis către autorităţile naţionale competente în conformitate cu termenele convenite, iar rapoartele periodice actualizate privind siguranţa (RPAS) vor fi depuse o dată la 3 ani. În plus, CHMP a analizat frecvenţa RPAS în cazul medicamentelor care conţin TCC pentru uz sistemic şi a solicitat depunerea RPAS o dată la trei ani (în loc de o dată la 13 ani, după cum se recomandă în prezent). Trebuie să se efectueze o monitorizare continuă a tuturor semnalelor privind siguranţa care sunt corelate cu aneuploidia (adică teratogenitate, toxicitate embrio-fetală / avort spontan, afectarea fertilităţii masculine şi cancer) şi trebuie să se urmărească raportarea sarcinilor pentru colectarea datelor structurate privind expunerea accidentală la medicament. În cadrul PMR trebuie furnizată o mostră a formularului de raportare a sarcinilor menţionate mai sus şi trebuie depus un raport al acestor date colectate în cadrul RPAS. În plus, CHMP a solicitat efectuarea unui studiu privind modul de utilizare a medicamentului (DUS) în scopul unei caracterizări mai bune a practicilor de prescriere a acestor medicamente în timpul utilizării clinice tipice de către grupurile reprezentative de medici prescriptori şi pentru evaluarea principalelor motive de prescriere. Acest studiu DUS trebuie efectuat pe o perioadă de trei ani. Protocolul studiului trebuie furnizat în cadrul PMR. În cele din urmă, materialul educaţional pentru medicii prescriptori şi pentru pacienţi care subliniază riscurile şi atenţionările referitoare la reacţiile de genotoxicitate va fi transmis şi autorităţilor naţionale competente în cadrul PMR. Raportul beneficiu-risc
Luând act de cele de mai sus, CHMP a concluzionat că raportul beneficiu-risc al medicamentelor care conţin TCC indicate ca tratament adjuvant al contracturilor musculare dureroase în patologia spinală acută la adulţi şi adolescenţi cu vârste de peste 16 ani rămâne favorabil sub rezerva restricţiilor, atenţionărilor, a altor modificări ale informaţiilor referitoare la produs, a activităţilor suplimentare de farmacovigilenţă şi a măsurilor convenite de reducere la minimum a riscurilor (MRMR).

19
Motivele menţinerii autorizaţiilor de punere pe piaţă
Întrucât
• Comitetul a analizat procedura în temeiul articolului 31 din Directiva 2001/83/CE pentru medicamentele care conţin tiocolchicozidă pentru uz sistemic (vezi anexa I).
• Comitetul a evaluat toate datele disponibile din studiile pre-clinice, din studiile clinice, din
studiile farmacoepidemiologice, din literatura de specialitate publicată şi din experienţa ulterioară punerii pe piaţă privind siguranţa medicamentelor care conţin tiocolchicozidă pentru uz sistemic, în ceea ce priveşte genotoxicitatea acesteia.
• Comitetul a considerat că medicamentele care conţin tiocolchicozidă pentru uz sistemic rămân un tratament adjuvant eficace al contracturilor musculare dureroase în patologia spinală acută. Cu toate acestea, în urma analizării riscurilor, medicamentele care conţin tiocolchicozidă pentru uz sistemic trebuie administrate numai la pacienţii cu vârste de peste 16 ani în tratarea afecţiunilor acute, cu durata tratamentului limitată la 7 zile consecutive (pentru medicamentele de uz oral) şi la 5 zile consecutive (pentru medicamentele administrate pe cale intramusculară). În această privinţă, CHMP a fost de părere că indicaţia în „boala Parkinson şi simptome parkinsoniene induse medicamentos, cu atenţie deosebită la sindromul dislexiei neurologice” trebuie eliminată deoarece aceasta este o afecţiune cronică care necesită o durată mai mare de tratament. Mărimea ambalajului trebuie adaptată la noile recomandări de tratament în funcţie de numărul de zile.
• De asemenea, Comitetul a considerat că medicamentele care conţin tiocolchicozidă pentru
uz sistemic trebuie contraindicate pe parcursul întregii perioade a sarcinii. De asemenea, aceste produse trebuie contraindicate la femeile cu potenţial fertil care nu utilizează medicamente contraceptive şi în timpul lactaţiei. De asemenea, CHMP a recomandat efectuarea unor modificări suplimentare ale informaţiilor referitoare la produs, inclusiv în informaţiile privind fertilitatea.
• De asemenea, CHMP a convenit asupra necesităţii unui plan de management al riscurilor
(PMR). În plus, toţi deţinătorii autorizaţiilor de punere pe piaţă ale acestor produse trebuie să depună RPAS odată la trei ani. Aceste RPAS trebuie să cuprindă un raport care prezintă date ale monitorizării continue a tuturor semnalelor privind siguranţa, corelate cu aneuploidia şi sarcina în cazul expunerii accidentale la medicament.
• Comitetul a concluzionat că era necesară luarea unor măsuri suplimentare de reducere la minimum a riscurilor, cum ar fi un studiu privind modul de utilizare a medicamentului pentru a caracteriza practicile de prescriere din timpul administrării clinice tipice, precum şi elaborarea materialelor educaţionale adecvate pentru pacienţi şi medicii prescriptori. Aceste măsuri urmează să fie incluse în PMR.
În consecinţă, Comitetul a concluzionat că raportul beneficiu-risc al medicamentelor care conţin tiocolchicozidă pentru uz sistemic ca tratament adjuvant al contracturilor musculare dureroase în patologia spinală acută la adulţi şi adolescenţi cu vârsta de peste 16 ani rămâne favorabil, sub rezerva restricţiilor, a atenţionărilor, a altor modificări ale informaţiilor referitoare la produs, a activităţilor suplimentare de farmacovigilenţă şi a măsurilor convenite de reducere la minimum a riscurilor.

20
Anexa III
Modificări aduse secţiunilor relevante ale rezumatului caracteristicilor produsului, etichetării şi prospectului

21
REZUMATUL CARACTERISTICILOR PRODUSULUI
[trebuie introdusă formularea de mai jos]
Acest medicament face obiectul unei monitorizări suplimentare. Acest lucru va permite identificarea rapidă de noi informaţii referitoare la siguranţă. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţii adverse suspectate. Vezi pct. 4.8 pentru modul de raportare a reacţiilor adverse. 4. DATE CLINICE 4.1 Indicaţii terapeutice [indicaţiile aprobate în prezent trebuie şterse şi înlocuite cu următoarele] Tratamentul adjuvant al contracturilor musculare dureroase în patologia spinală acută la adulţi şi adolescenţi cu vârsta peste 16 ani. 4.2 Doze şi mod de administrare [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] Doze o Pentru forma orală de 4 mg şi 8 mg: Doza recomandată şi maximă este de 8 mg la fiecare 12 ore (adică, 16 mg pe zi). Durata tratamentului este limitată la 7 zile consecutive. o Pentru forma i.m.: Doza recomandată şi maximă este de 4 mg la fiecare 12 ore (adică, 8 mg pe zi). Durata tratamentului este limitată la 5 zile consecutive. o Atât pentru forma orală, cât şi pentru forma i.m.: Trebuie să se evite dozele care depăşesc doza recomandată sau utilizarea de lungă durată (vezi pct. 4.4). Copii şi adolescenţi <Numele inventat> nu trebuie utilizat la copii şi adolescenţi cu vârsta sub 16 ani, din motive de siguranţă (vezi pct. 5.3). Mod de administrare [A se completa la nivel naţional] 4.3 Contraindicaţii [trebuie introdusă formularea de mai jos] Tiocolchicozida nu trebuie utilizată - la pacienţii hipersensibili la substanţa activă sau la oricare dintre excipienţii enumeraţi la pct. 6.1 - pe întreaga durată a perioadei de sarcină - în timpul alăptării - la femeile aflate la vârsta fertilă care nu utilizează măsuri contraceptive. 4.4 Atenţionări şi precauţii speciale pentru utilizare [trebuie introdusă formularea de mai jos] […]

22
Studiile preclinice au arătat că unul dintre metaboliţii tiocolchicozidei (SL59.0955) a indus aneuploidie (adică, număr inegal de cromozomi în celulele divizate) la concentraţii apropiate de expunerea la om, aceasta fiind observată la doze de 8 mg administrate de două ori pe zi, pe cale orală (vezi pct. 5.3). Aneuploidia este considerată un factor de risc pentru teratogenicitate, toxicitate embrionară şi fetală, avort spontan şi alterarea fertilităţii masculine, precum şi un potenţial factor de risc pentru cancer. Ca măsură de precauţie, trebuie să se evite utilizarea medicamentului la doze care depăşesc doza recomandată sau utilizarea de lungă durată (vezi pct. 4.2). Pacienţii trebuie informaţi atent cu privire la riscul potenţial al unei posibile sarcini şi la măsurile contraceptive eficace care trebuie respectate. 4.6 Fertilitatea, sarcina şi alăptarea [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] […] Sarcina Datele privind utilizarea tiocolchicozidei la femeile gravide sunt limitate. Prin urmare, nu se cunosc pericolele potenţiale pentru embrion şi făt. Studiile la animale au evidenţiat efecte teratogene (vezi pct. 5.3). <Numele inventat> este contraindicat în timpul sarcinii şi la femei aflate la vârsta fertilă care nu utilizează măsuri contraceptive (vezi pct. 4.3). Alăptarea Întrucât este excretată în laptele matern, utilizarea tiocolchicozidei este contraindicată în timpul alăptării (vezi pct. 4.3). Fertilitatea În cadrul unui studiu privind fertilitatea efectuat la şobolani, nu s-a observat nicio alterare a fertilităţii la doze de până la 12 mg/kg, adică la nivelurile de doză care nu induc niciun efect clinic. Tiocolchicozida şi metaboliţii săi exercită activitate aneugenică la diferite niveluri de concentraţie, ceea ce reprezintă un factor de risc pentru afectarea fertilităţii la om (vezi pct. 5.3). 4.8 Reacţii adverse […] [trebuie introdusă formularea de mai jos] Raportarea reacţiilor adverse suspectate Raportarea reacţiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată prin intermediul sistemului naţional de raportare, aşa cum este menţionat în Anexa V*. [*Pentru materialul tipărit, vă rugăm să consultaţi ghidul şablonului QRD adnotat.] […] 5. PROPRIETĂŢI FARMACOLOGICE 5.2 Proprietăţi farmacocinetice [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] Absorbţie

23
- După administrarea i.m., nivelul Cmax al tiocolchicozidei survine în 30 de minute şi atinge valori de 113 ng/ml după o doză de 4 mg şi de 175 mg/ml după o doză de 8 mg. Valorile corespunzătoare ale ASC sunt de 283 şi, respectiv, 417 ng.h/ml. Metabolitul activ din punct de vedere farmacologic SL18.0740 se observă, de asemenea, la concentraţii scăzute, cu un nivel Cmax de 11,7 ng/ml, care survine la 5 ore după doză şi o valoare ASC de 83 ng.h/ml. Nu sunt disponibile date pentru metabolitul inactiv SL59.0955. - După administrarea orală, nu se detectează nicio cantitate de tiocolchicozidă în plasmă. Se observă numai doi metaboliţi: Metabolitul activ din punct de vedere farmacologic SL18.0740 şi un metabolit inactiv SL59.0955. Pentru ambii metaboliţi, concentraţiile plasmatice maxime survin la 1 oră după administrarea tiocolchicozidei. După administarea unei doze orale unice de 8 mg de tiocolchicozidă, valorile Cmax şi ASC ale SL18.0740 sunt de aproximativ 60 ng/ml şi, respectiv, 130 ng.h/ml. Pentru SL59.0955, aceste valori sunt mult mai scăzute: Cmax este de aproximativ 13 ng/ml, iar ASC variază între 15,5 ng.h/ml (până la 3 ore) şi 39,7 ng.h/ml (până la 24 de ore). Distribuţie Volumul aparent de distribuţie al tiocolchicozidei este estimat la aproximativ 42,7 l după administarea IM a unei dozei de 8 mg. Nu sunt disponibile date pentru ambii metaboliţi. Metabolizare După administrarea orală, tiocolchicozida este metabolizată mai întâi în aglicon 3-demetiltiocolchicină sau SL59.0955. Această etapă survine în principal prin metabolizarea intestinală, explicând lipsa de tiocolchicozidă nemodificată circulantă ca urmare a utilizării acestei căi de administrare. SL59.0955 este apoi glucuroconjugat în SL18.0740, care are o activitate farmacologică echipotenţială celei a tiocolchicozidei, sprijinind astfel activitatea farmacologică după administrarea orală a tiocolchicozidei. SL59.0955 este, de asemenea, demetilat în didemetil-tiocolchicină. Eliminare - După administrarea i.m., timpul de înjumătăţire plasmatică prin eliminare aparent al tiocolchicozidei este de 1,5 ore, iar clearance-ul plasmatic este de 19,2 l/h. - După administrarea orală, radioactivitatea totală este în principal excretată în materiile fecale (79%), în timp ce excreţia pe cale urinară reprezintă numai 20%. Nu se excretă tiocolchicozidă nemodificată nici în urină, nici în materiile fecale. SL18.0740 şi SL59.0955 se găsesc în urină şi materiile fecale, în timp ce didemetil-tiocolchicina este recuperată numai în materiile fecale. După administrarea orală a tiocolchicozidei, metabolitul SL18.0740 este eliminat cu un timp de înjumătăţire plasmatică prin eliminare aparent care variază între 3,2 şi 7 ore, iar metabolitul SL59.0955 are un timp de înjumătăţire plasmatică prin eliminare mediu de 0,8 ore. 5.3 Date preclinice de siguranţă [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] Profilul tiocolchicozidei a fost evaluat in vitro şi in vivo după administrarea parenterală şi orală. Tiocolchicozida a fost bine tolerată după administrarea orală în decursul perioadelor de până la 6 luni, atât la şobolani, cât şi la primate neumane, atunci când aceasta a fost administrată în doze repetate mai mici sau egale cu 2 mg/kg/zi la şobolani şi mai mici sau egale cu 2,5 mg/kg/zi la primate neumane, precum şi după administrarea pe cale intramusculară la primate, în doze repetate de până la 0,5 mg/kg/zi timp de 4 săptămâni. La doze ridicate, tiocolchicozida a indus emeză la câini, diaree la şobolani şi convulsii atât la rozătoare, cât şi la nerozătoare după administrarea acută pe cale orală. După administrarea repetată, tiocolchicozida a indus tulburări gastro-intestinale (enterită, emeză) la utilizarea căii orale şi emeză la utilizarea căii i.m.. Tiocolchicozida în sine nu a indus mutaţie genetică la bacterii (testul Ames), deteriorare cromozomială in vitro (testul de anomalii cromozomiale pe limfocite umane) şi deteriorare cromozomială in vivo (testul in vivo de micronucleu pe măduva osoasă a şoarecilor, cu administrare intraperitoneală). Metabolitul glucuroconjugat major SL18.0740 nu a indus mutaţie genetică la bacterii (testul Ames); cu toate acestea a indus deteriorare cromozomială in vitro (testul in vitro de micronucleu pe limfocite umane) şi deteriorare cromozomială in vivo (testul in vivo de micronucleu pe măduva osoasă a şoarecilor, cu administrare orală). Micronucleii au rezultat preponderent din pierderea cromozomială (centromeri cu micronuclei pozitivi după colorarea FISH a centromerilor), ceea ce sugerează proprietăţi aneugenice. Efectul aneugenic al SL18.0740 a fost observat la concentraţii, în cadrul testului in vitro, şi la expuneri plasmatice ASC, în cadrul testului in vivo, mai ridicate (de peste 10 ori pe baza ASC) decât cele observate în plasma umană la doze terapeutice.

24
Metabolitul aglicon (3-demetiltiocolchicină-SL59.0955), format în principal după administrarea orală, a indus deteriorare cromozomială in vitro (testul in vitro de micronucleu pe limfocite umane) şi deteriorare cromozomială in vivo (testul in vivo de micronucleu oral pe măduva osoasă a şobolanilor, cu administrare orală. Micronucleii au rezultat preponderent din pierderea cromozomială (centromeri cu micronuclei pozitivi după colorarea FISH sau CREST a centromerilor), ceea ce sugerează proprietăţi aneugenice. Efectul aneugenic al SL59.0955 a fost observat la concentraţii, în cadrul testului in vitro, şi la expuneri, în cadrul testului in vivo, apropiate de cele observate în plasma umană la doze terapeutice de 8 mg administrate de două ori pe zi, pe cale orală. Efectul aneugenic în celulele divizate poate duce la celule aneuploide. Aneuploidia reprezintă o modificare a numărului de cromozomi şi pierderea heterozigozităţii, fenomen care este recunoscut drept un factor de risc pentru teratogenicitate, toxicitate embrionară/avort spontan, alterarea fertilităţii masculine, atunci când afectează celulele germinale, precum şi un factor de risc potenţial pentru cancer, atunci când afectează celulele somatice. Prezenţa metabolitului aglicon (3-demetiltiocolchicină-SL59.0955) după administrarea intramusculară nu a fost evaluată niciodată; prin urmare, formarea acestuia utilizând această cale de administrare nu poate fi exclusă. La şobolani, o doză orală de 12 mg/kg/zi de tiocolchicozidă a cauzat malformaţii majore, precum şi toxicitate fetală (întârzieri de creştere, moarte embrionară, alterarea ratei de distribuţie a sexelor). Doza fără efect toxic a fost de 3 mg/kg/zi. La iepuri, tiocolchicozida a demonstrat materno-toxicitate la doze începând de la 24 mg/kg/zi. Mai mult decât atât, au fost observate anomalii minore (coaste supranumerare, întârzieri de osificare). În cadrul unui studiu privind fertilitatea efectuat pe şobolani, nu s-a observat nicio alterare a fertilităţii la doze de până la 12 mg/kg/zi, adică la nivelurile de doză care nu induc niciun efect clinic. Tiocolchicozida şi metaboliţii săi exercită activitate aneugenică la diferite niveluri de concentraţie, fenomen care este recunoscut drept un factor de risc pentru alterarea fertilităţii la om. Potenţialul carcinogenic nu a fost evaluat. 6.5 Natura şi conţinutul ambalajului <şi echipamente speciale pentru utilizare, administrare sau implantare> [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] 30 comprimate/capsule pentru doza de 4 mg şi 14 comprimate/capsule pentru doza de 8 mg. 10 flacoane / fiole pentru doza de 4 mg / 2 ml.

25
ETICHETAREA
INFORMAŢII CARE TREBUIE SĂ APARĂ PE AMBALAJUL SECUNDAR Cutie pentru capsule/comprimate/comprimate orodispersabile şi soluţie injectabilă 4. FORMA FARMACEUTICĂ ŞI CONŢINUTUL [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea]
4 mg [până la 30] capsule [până la 30] comprimate
8 mg [până la 14] capsule [până la 14] comprimate orodispersabile
4 mg/2 ml [până la 10] flacoane/fiole

26
PROSPECTUL [trebuie introdusă formularea de mai jos]
Acest medicament face obiectul unei monitorizări suplimentare. Acest lucru va permite identificarea rapidă de noi informaţii referitoare la siguranţă. Puteţi să fiţi de ajutor raportând orice reacţii adverse pe care le puteţi avea. Vezi ultima parte de la pct. 4 pentru modul de raportare a reacţiilor adverse. […]
Prospect
Prospect: Informaţii pentru pacient 1. Ce este X şi pentru ce se utilizează [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] Acest medicament este un relaxant muscular. Acesta se utilizează la adulţi şi adolescenţi cu vârsta peste 16 ani, ca tratament adjuvant pentru contracţiile musculare dureroase. Acesta trebuie să se utilizeze pentru stări acute legate de coloana vertebrală. 2. Ce trebuie să ştiţi înainte să luaţi X [trebuie introdusă formularea de mai jos] Nu luaţi X: - dacă sunteţi alergic la tiocolchicozidă sau la oricare dintre celelalte componente ale acestui medicament (enumerate la pct. 6) - sunteţi gravidă, aţi putea rămâne gravidă sau credeţi că aţi putea fi gravidă - sunteţi o femeie aflată la vârsta fertilă care nu utilizează măsuri contraceptive - alăptaţi Atenţionări şi precauţii […] Respectaţi cu stricteţe dozele şi durata tratamentului, care sunt prezentate în detaliu la pct. 3. Nu trebuie să utilizaţi acest medicament în doze ridicate sau timp de mai mult de 7 zile (pentru formele orale)/5 zile (pentru formele i.m.). Aceasta deoarece unul dintre produsele formate în corpul dumneavoastră atunci când luaţi tiocolchicozidă în doze ridicate poate cauza deteriorări ale unor celule (număr anormal de cromozomi). Acest efect a fost demonstrat în cadrul studiilor pe animale şi al studiilor de laborator. La om, acest tip de deteriorare a celulelor reprezintă un factor de risc pentru cancer, vătămarea fătului şi alterarea fertilităţii masculine. Vă rugăm să vă adresaţi medicului dumneavoastră dacă aveţi întrebări suplimentare. Medicul dumneavoastră vă va informa cu privire la măsurile legate de contracepţia eficace şi la potenţialul risc al unei sarcini. Copii şi adolescenţi Nu administraţi acest medicament copiilor şi adolescenţilor cu vârsta sub 16 ani din motive de siguranţă. Sarcina, alăptarea şi fertilitatea [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] Nu luaţi acest medicament dacă: - sunteţi gravidă, aţi putea rămâne gravidă sau credeţi că aţi putea fi gravidă - sunteţi o femeie aflată la vârsta fertilă care nu utilizează măsuri contraceptive

27
Aceasta deoarece acest medicament poate dăuna fătului. Nu luaţi acest medicament dacă alăptaţi. Aceasta deoarece medicamentul este excretat în laptele matern. Acest medicament poate cauza probleme de fertilitate masculină din cauza potenţialei deteriorări a celulelor spermatice (număr anormal de cromozomi). Această afirmaţie se bazează pe studii de laborator (vezi pct. 2 „Atenţionări şi precauţii”). 3. Cum să luaţi X [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] Luaţi întotdeauna acest medicament exact aşa cum v-a spus medicul sau farmacistul. Discutaţi cu medicul dumneavoastră sau cu farmacistul dacă nu sunteţi sigur. o Pentru forma orală de 4 mg şi 8 mg: Doza recomandată şi maximă este de 8 mg la fiecare 12 ore (adică, 16 mg pe zi). Durata tratamentului este limitată la 7 zile consecutive. o Pentru forma intramusculară: Doza recomandată şi maximă este de 4 mg la fiecare 12 ore (adică, 8 mg pe zi). Durata tratamentului este limitată la 5 zile consecutive. o Atât pentru forma orală, cât şi pentru forma intramusculară: Nu depăşiţi dozele recomandate şi durata tratamentului. Acest medicament nu trebuie utilizat pentru tratamentul pe termen lung (vezi pct. 2 „Atenţionări şi precauţii”). Utilizarea la copii şi adolescenţi Nu administraţi acest medicament copiilor şi adolescenţilor cu vârsta sub 16 ani din motive de siguranţă. Dacă luaţi mai mult X decât trebuie Dacă luaţi accidental mai mult X decât trebuie, adresaţi-vă medicului dumneavoastră, farmacistului sau asistentei medicale. Dacă uitaţi să luaţi X Nu luaţi o doză dublă pentru a compensa doza uitată. Dacă aveţi orice întrebări suplimentare cu privire la acest medicament, adresaţi-vă medicului dumneavoastră, farmacistului sau asistentei medicale. 4. Reacţii adverse posibile [trebuie introdusă această formulare] Ca toate medicamentele, acest medicament poate provoca reacţii adverse, cu toate că nu apar la toate persoanele. […] [trebuie introdusă formularea de mai jos]
Raportarea reacţiilor adverse Dacă manifestaţi orice reacţii adverse, adresaţi-vă medicului dumneavoastră, farmacistului sau asistentei medicale. Acestea includ orice reacţii adverse nemenţionate în acest prospect. De asemenea, puteţi raporta reacţiile adverse direct prin intermediul {sistemului naţional de raportare, aşa cum este menţionat în Anexa V}*. Raportând reacţiile adverse, puteţi contribui la furnizarea de informaţii suplimentare privind siguranţa acestui medicament. [*Pentru materialul tipărit, vă rugăm să consultaţi ghidul şablonului QRD adnotat.]
6. Conţinutul ambalajului şi alte informaţii [formularea aprobată în prezent trebuie ştearsă şi înlocuită cu următoarea] 30 comprimate/capsule pentru doza de 4 mg şi 14 comprimate/capsule pentru doza de 8 mg. 10 flacoane / fiole pentru doza de 4 mg / 2 ml.

28
Anexa IV
Condiţii pentru autorizaţiile de punere pe piaţă
29
Condiţii pentru autorizaţiile de punere pe piaţă
Autorităţile naţionale competente ale statului (statelor) membru (membre) coordonate de statul (statele) membru (membre) de referinţă, dacă este cazul, se asigură că următoarele condiţii sunt îndeplinite de către deţinătorul (deţinătorii) autorizaţiei (autorizaţiilor) de punere pe piaţă (DAPP):
Condiţii Data
Deţinătorii autorizaţiilor de punere pe piaţă trebuie să difuzeze comunicarea directă către profesioniştii din domeniul sănătăţii (DHCP) prin cooperare cu autorităţile naţionale competente (ANC) în conformitate cu planul de acţiune aprobat de CHMP.
În termen de 30 de zile de la decizia CE
Deţinătorii autorizaţiilor de punere pe piaţă trebuie să depună un plan de management al riscurilor (inclusiv rezumatul studiului DUS şi materialele educaţionale, menţionate şi mai jos) în format UE.
În termen de 2 luni de la decizia CE
Tiocolchicozida face parte dintr-un proiect de sincronizare a RPAS al directorilor autorităţilor de reglementare în domeniul medicamentelor.
Deţinătorii autorizaţiilor de punere pe piaţă trebuie să depună următoarea raportare RPAS până la data de:
4 iulie 2015
În cadrul prezentării planului de management al riscurilor, deţinătorii autorizaţiilor de punere pe piaţă trebuie să furnizeze un protocol al studiului privind modul de utilizare a medicamentului pentru a caracteriza practicile de prescriere a medicamentelor din timpul administrării clinice tipice de către grupurile reprezentative de medici prescriptori şi pentru a evalua motivele principale ale prescrierii. Raportul final al studiului trebuie prezentat până în luna:
noiembrie 2017
În cadrul prezentării planului de management al riscurilor, deţinătorii autorizaţiilor de punere pe piaţă trebuie să furnizeze materiale educaţionale pentru medicii prescriptori şi pentru pacienţi. În acestea vor fi subliniate riscurile şi atenţionările privind reacţiile de genotoxicitate.
În termen de 2 luni de la decizia CE